Réaction chimique
Une réaction Chimique est un processus qui conduit à la transformation Chimique d’un ensemble de substances chimiques en un autre. [1] Classiquement, les réactions chimiques englobent des changements qui n’impliquent que les positions des électrons dans la formation et la rupture des liaisons chimiques entre les atomes , sans changement pour les noyaux (pas de changement pour les éléments présents), et peuvent souvent être décrites par une réaction Chimique . équation . La chimie nucléaire est une sous-discipline de la chimie qui implique les réactions chimiques d’ éléments instables et éléments radioactifs où des changements électroniques et nucléaires peuvent se produire.
 Une réaction aluminothermique utilisant de l’oxyde de fer (III). Les étincelles qui s’envolent vers l’extérieur sont des globules de fer en fusion traînant de la fumée dans leur sillage.
Une réaction aluminothermique utilisant de l’oxyde de fer (III). Les étincelles qui s’envolent vers l’extérieur sont des globules de fer en fusion traînant de la fumée dans leur sillage.
La substance (ou les substances) initialement impliquée dans une réaction Chimique sont appelées réactifs ou réactifs . Les réactions chimiques sont généralement caractérisées par un changement Chimique , et elles donnent un ou plusieurs produits , qui ont généralement des propriétés différentes de celles des réactifs. Les réactions consistent souvent en une séquence de sous-étapes individuelles, appelées réactions élémentaires , et les informations sur le déroulement précis de l’action font partie du mécanisme de réaction . Les réactions chimiques sont décrites avec des équations chimiques , qui présentent symboliquement les matières premières, les produits finaux et parfois les produits intermédiaires et les conditions de réaction.
Les réactions chimiques se produisent à une vitesse de réaction caractéristique à une température et une concentration Chimique données. En règle générale, les vitesses de réaction augmentent avec l’augmentation de la température car il y a plus d’énergie thermique disponible pour atteindre l’énergie d’activation nécessaire pour rompre les liaisons entre les atomes.
Les réactions peuvent se dérouler dans le sens direct ou inverse jusqu’à ce qu’elles se terminent ou atteignent l’équilibre . Les réactions qui se déroulent dans le sens direct pour approcher l’équilibre sont souvent décrites comme spontanées , ne nécessitant aucun apport d’énergie libre pour avancer. Les réactions non spontanées nécessitent un apport d’énergie libre pour avancer (par exemple, charger une batterie en appliquant une source d’alimentation électrique externe ou la photosynthèse entraînée par l’absorption du rayonnement électromagnétique sous forme de lumière solaire).
Une réaction peut être classée comme redox dans laquelle l’Oxydation et la réduction se produisent ou non redox dans laquelle il n’y a pas d’Oxydation et de réduction. La plupart des réactions redox simples peuvent être classées en réactions de combinaison, de décomposition ou de déplacement unique.
Différentes réactions chimiques sont utilisées lors de la synthèse Chimique afin d’obtenir un produit souhaité. En biochimie , une série consécutive de réactions chimiques (où le produit d’une réaction est le Réactif de la réaction suivante) forment des voies métaboliques . Ces réactions sont souvent catalysées par des enzymes protéiques . Les enzymes augmentent les taux de réactions biochimiques, de sorte que des synthèses métaboliques et des décompositions impossibles dans des conditions ordinaires peuvent se produire aux températures et concentrations présentes dans une cellule .
Le concept général de réaction Chimique a été étendu aux réactions entre des entités plus petites que les atomes, y compris les Réactions nucléaires , les désintégrations radioactives et les réactions entre particules élémentaires , telles que décrites par la théorie quantique des champs .
Histoire
 Antoine Lavoisier a développé la théorie de la combustion en tant que réaction Chimique avec l’oxygène.
Antoine Lavoisier a développé la théorie de la combustion en tant que réaction Chimique avec l’oxygène.
Les réactions chimiques telles que la combustion dans le feu, la fermentation et la réduction des minerais en métaux étaient connues depuis l’Antiquité. Les théories initiales de la transformation des matériaux ont été développées par des philosophes grecs, comme la théorie des quatre éléments d’ Empédocle déclarant que toute substance est composée des quatre éléments de base – le feu, l’eau, l’air et la terre. Au Moyen Age , les transformations chimiques étaient étudiées par les alchimistes . Ils ont tenté, en particulier, de convertir le plomb en or , pour cela ils ont utilisé des réactions du plomb et des alliages plomb-cuivre avec le soufre . [2]
La production artificielle de substances chimiques était déjà un objectif central pour les alchimistes médiévaux. [3] Les exemples incluent la synthèse de chlorure d’ammonium à partir de substances organiques comme décrit dans les travaux (vers 850–950) attribués à Jābir ibn Ḥayyān , [4] ou la production d’ Acides minéraux tels que les acides sulfurique et nitrique par des alchimistes ultérieurs, à partir de c. 1300. [5] La production d’Acides minéraux impliquait le chauffage de minéraux sulfates et nitrates tels que le sulfate de cuivre , l’ alun et le salpêtre . Au 17ème siècle,Johann Rudolph Glauber a produit de l’acide chlorhydrique et du sulfate de sodium en faisant réagir de l’acide sulfurique et du chlorure de sodium . Avec le développement du procédé à chambre de plomb en 1746 et du procédé Leblanc , permettant la production à grande échelle d’acide sulfurique et de carbonate de sodium , respectivement, les réactions chimiques se sont mises en œuvre dans l’industrie. Une optimisation supplémentaire de la technologie de l’acide sulfurique a abouti au processus de contact dans les années 1880, [6] et le processus Haber a été développé en 1909–1910 pour la synthèse d’ ammoniac . [7]
À partir du XVIe siècle, des chercheurs dont Jan Baptist van Helmont , Robert Boyle et Isaac Newton ont tenté d’établir des théories sur les transformations chimiques observées expérimentalement. La théorie du phlogistique a été proposée en 1667 par Johann Joachim Becher . Elle postulait l’existence d’un élément semblable au feu appelé “phlogiston”, qui était contenu dans des corps combustibles et libéré lors de la combustion . Cela s’est avéré faux en 1785 par Antoine Lavoisier qui a trouvé la bonne explication de la combustion comme réaction avec l’oxygène de l’air. [8]
Joseph Louis Gay-Lussac a reconnu en 1808 que les gaz réagissent toujours dans un certain rapport entre eux. Sur la base de cette idée et de la théorie atomique de John Dalton , Joseph Proust avait développé la loi des proportions définies , qui a ensuite abouti aux concepts de stoechiométrie et d ‘ équations chimiques . [9]
En ce qui concerne la chimie organique , on a longtemps cru que les composés obtenus à partir d’organismes vivants étaient trop complexes pour être obtenus par synthèse . Selon le concept de vitalisme , la matière organique était dotée d’une “force vitale” et se distinguait des matières inorganiques. Cette séparation a cependant pris fin avec la synthèse de l’urée à partir de précurseurs inorganiques par Friedrich Wöhler en 1828. D’autres chimistes qui ont apporté des contributions majeures à la chimie organique incluent Alexander William Williamson avec sa synthèse des éthers et Christopher Kelk Ingold , qui, parmi de nombreuses découvertes, a établi le mécanismes deréactions de substitution .
Les caractéristiques
| Cette section a besoin d’être agrandie . Vous pouvez aider en y ajoutant . ( Novembre 2020 ) |
Les caractéristiques générales des réactions chimiques sont :
- Évolution d’un gaz
- Formation d’un précipité
- Changement de température
- Changement d’ état
Équations
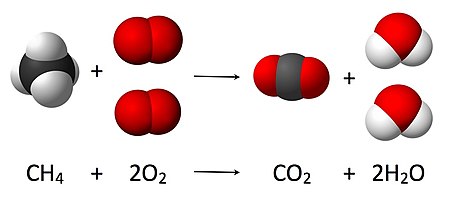 Comme le montre l’équation CH 4 + 2O 2 → CO 2 + 2 H 2 O , un coefficient de 2 doit être placé avant l’ oxygène gazeux côté réactifs et avant l’ eau côté produits pour que, conformément à la loi de conservation de la masse, la quantité de chaque élément ne change pas au cours de la réaction
Comme le montre l’équation CH 4 + 2O 2 → CO 2 + 2 H 2 O , un coefficient de 2 doit être placé avant l’ oxygène gazeux côté réactifs et avant l’ eau côté produits pour que, conformément à la loi de conservation de la masse, la quantité de chaque élément ne change pas au cours de la réaction
Les équations chimiques sont utilisées pour illustrer graphiquement les réactions chimiques. Ils sont constitués des formules chimiques ou structurales des réactifs à gauche et de celles des produits à droite. Ils sont séparés par une flèche (→) qui indique le sens et le type de réaction ; la flèche est lue comme le mot “rend”. [10] La pointe de la flèche pointe dans la direction dans laquelle la réaction se déroule. Une double flèche (⇌) pointant dans des directions opposées est utilisée pour les réactions d’équilibre . Les équations doivent être équilibrées en fonction de la stoechiométrie, le nombre d’atomes de chaque espèce doit être le même des deux côtés de l’équation. Ceci est réalisé en mettant à l’échelle le nombre de molécules impliquées (A, B, C et D dans un exemple schématique ci-dessous) par les nombres entiers appropriés a, b, c et d . [11]
une UNE + b B → c C + ré D
Des réactions plus élaborées sont représentées par des schémas de réaction qui, en plus des matières premières et des produits, présentent des intermédiaires ou des états de transition importants . En outre, certains ajouts relativement mineurs à la réaction peuvent être indiqués au-dessus de la flèche de réaction ; des exemples de tels ajouts sont l’eau, la chaleur, l’éclairage, un Catalyseur , etc. De même, certains produits mineurs peuvent être placés sous la flèche, souvent avec un signe moins.
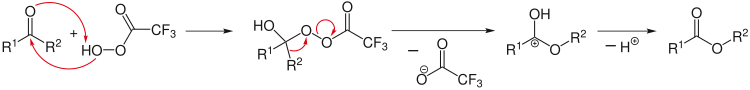 Un exemple de réaction organique : Oxydation des cétones en esters avec un acide peroxycarboxylique
Un exemple de réaction organique : Oxydation des cétones en esters avec un acide peroxycarboxylique
L’analyse rétrosynthétique peut être appliquée pour concevoir une Réaction de synthèse complexe. Ici, l’analyse part des produits, par exemple en séparant des liaisons chimiques sélectionnées, pour arriver à des réactifs initiaux plausibles. Une flèche spéciale (⇒) est utilisée dans les réactions rétro. [12]
Réactions élémentaires
La réaction élémentaire est la plus petite division dans laquelle une réaction Chimique peut être décomposée, elle n’a pas de produits intermédiaires. [13] La plupart des réactions observées expérimentalement sont construites à partir de nombreuses réactions élémentaires qui se produisent en parallèle ou séquentiellement. La séquence réelle des réactions élémentaires individuelles est connue sous le nom de mécanisme de réaction . Une réaction élémentaire implique quelques molécules, généralement une ou deux, en raison de la faible probabilité que plusieurs molécules se rencontrent à un certain moment. [14]
 Isomérisation de l’ azobenzène , induite par la lumière (hν) ou la chaleur (Δ)
Isomérisation de l’ azobenzène , induite par la lumière (hν) ou la chaleur (Δ)
Les réactions élémentaires les plus importantes sont les réactions unimoléculaires et bimoléculaires. Une seule molécule est impliquée dans une réaction unimoléculaire ; il est transformé par une isomérisation ou une dissociation en une ou plusieurs autres molécules. De telles réactions nécessitent l’ajout d’énergie sous forme de chaleur ou de lumière. Un exemple typique de réaction unimoléculaire est l’ isomérisation cis-trans , dans laquelle la forme cis d’un composé se transforme en forme trans ou vice versa. [15]
Dans une réaction de dissociation typique , une liaison dans une molécule se divise ( rupture ) résultant en deux fragments moléculaires. Le clivage peut être homolytique ou hétérolytique . Dans le premier cas, la liaison est divisée de sorte que chaque produit retient un électron et devient un radical neutre . Dans le second cas, les deux électrons de la liaison Chimique restent avec l’un des produits, ce qui donne des ions chargés . La dissociation joue un rôle important dans le déclenchement des réactions en chaîne , telles que les réactions hydrogène-oxygène ou de polymérisation .
UN B ⟶ UN + B {displaystyle {ce {AB -> A + B}}} 
Pour les réactions bimoléculaires , deux molécules entrent en collision et réagissent l’une avec l’autre. Leur fusion est appelée synthèse Chimique ou réaction d’addition .
UN + B ⟶ UN B {displaystyle {ce {A + B -> AB}}} 
Une autre possibilité est que seule une partie d’une molécule soit transférée à l’autre molécule. Ce type de réaction se produit, par exemple, dans les réactions redox et acide-base. Dans les réactions redox, la particule transférée est un électron, alors que dans les réactions acide-base, c’est un proton. Ce type de réaction est également appelé métathèse .
HA + B ⟶ A + HB {displaystyle {ce {HA + B -> A + HB}}} 
par exemple
NaCl + AgNO 3 ⟶ NaNO 3 + AgCl ↓ {displaystyle {ce {NaCl + AgNO3 -> NaNO3 + AgCl(v)}}} 
Équilibre Chimique
La plupart des réactions chimiques sont réversibles ; c’est-à-dire qu’ils peuvent fonctionner et fonctionnent dans les deux sens. Les réactions directes et inverses sont en concurrence les unes avec les autres et diffèrent dans les taux de réaction . Ces vitesses dépendent de la concentration et changent donc avec le temps de la réaction : la vitesse inverse augmente progressivement et devient égale à la vitesse de la réaction directe, établissant ce que l’on appelle l’équilibre Chimique. Le temps nécessaire pour atteindre l’équilibre dépend de paramètres tels que la température, la pression et les matériaux impliqués, et est déterminé par l’ énergie libre minimale . A l’équilibre, l’ énergie libre de Gibbs doit être nulle. La dépendance à la pression peut être expliquée par le principe de Le Chatelier. Par exemple, une augmentation de la pression due à la diminution du volume provoque le déplacement de la réaction vers le côté avec le moins de moles de gaz. [16]
Le rendement de la réaction se stabilise à l’équilibre, mais peut être augmenté en retirant le produit du mélange réactionnel ou modifié en augmentant la température ou la pression. Un changement dans les concentrations des réactifs n’affecte pas la constante d’équilibre, mais affecte la position d’équilibre.
Thermodynamique
Les réactions chimiques sont déterminées par les lois de la thermodynamique . Les réactions peuvent se dérouler d’elles-mêmes si elles sont exergoniques , c’est-à-dire si elles libèrent de l’énergie. L’énergie libre associée de la réaction est composée de deux grandeurs thermodynamiques différentes, l’ enthalpie et l’ entropie : [17]
Δ G = Δ H − T ⋅ Δ S {displaystyle Delta G=Delta HTcdot Delta S} 
Les réactions peuvent être exothermiques , où ΔH est négatif et de l’énergie est libérée. Des exemples typiques de réactions exothermiques sont la précipitation et la cristallisation , dans lesquelles des solides ordonnés sont formés à partir de phases gazeuses ou liquides désordonnées. En revanche, en endothermieréactions, la chaleur est consommée de l’environnement. Cela peut se produire en augmentant l’entropie du système, souvent par la formation de produits de réaction gazeux, qui ont une entropie élevée. Comme l’entropie augmente avec la température, de nombreuses réactions endothermiques ont lieu de préférence à des températures élevées. Au contraire, de nombreuses réactions exothermiques telles que la cristallisation se produisent à basse température. Les changements de température peuvent parfois inverser le signe de l’enthalpie d’une réaction, comme pour la réduction du monoxyde de carbone du dioxyde de molybdène :
2 CO ( g ) + MoO 2 ( s ) ⟶ 2 CO 2 ( g ) + Mo ( s ) {displaystyle {ce {2CO(g) + MoO2(s) -> 2CO2(g) + Mo(s)}}} 

Cette réaction pour former du dioxyde de carbone et du molybdène est Endothermique à basse température, devenant moins avec l’augmentation de la température. [18] ΔH° est nul à1855 K , et la réaction devient exothermique au-dessus de cette température.
Les changements de température peuvent également inverser la tendance de direction d’une réaction. Par exemple, la Réaction de changement de gaz à l’eau
CO ( g ) + H 2 O ( v ) ↽ − − ⇀ CO 2 ( g ) + H 2 ( g ) {displaystyle {ce {CO(g) + H2O({v}) <=> CO2(g) + H2(g)}}} 
est favorisé par les basses températures, mais son inverse est favorisé par les hautes températures. Le changement de tendance dans le sens de la réaction se produit à1100K . [18]
Les réactions peuvent également être caractérisées par l’ énergie interne qui tient compte des changements d’entropie, de volume et de potentiel Chimique . Cette dernière dépend, entre autres, des activités des substances impliquées. [19]
d U = T ⋅ d S − p ⋅ d V + μ ⋅ d n {displaystyle {d}U=Tcdot {d}Spcdot {d}V+mu cdot {d}n} 
Cinétique
La vitesse à laquelle les réactions ont lieu est étudiée par la cinétique de réaction . Le taux dépend de divers paramètres, tels que :
- Les concentrations de réactifs , qui accélèrent généralement la réaction si elles sont augmentées par une augmentation des collisions par unité de temps. Certaines réactions, cependant, ont des taux qui sont indépendants des concentrations de réactifs. C’est ce qu’on appelle des réactions d’ordre zéro .
- Surface disponible pour le contact entre les réactifs, notamment solides dans les systèmes hétérogènes. Des surfaces plus grandes conduisent à des taux de réaction plus élevés.
- Pression – l’augmentation de la pression diminue le volume entre les molécules et augmente donc la fréquence des collisions entre les molécules.
- L’énergie d’activation , qui est définie comme la quantité d’énergie nécessaire pour que la réaction démarre et se poursuive spontanément. Une énergie d’activation plus élevée implique que les réactifs ont besoin de plus d’énergie pour démarrer qu’une réaction avec une énergie d’activation plus faible.
- Température , qui accélère les réactions si elle est élevée, car une température plus élevée augmente l’énergie des molécules, créant plus de collisions par unité de temps,
- La présence ou l’absence d’un Catalyseur . Les catalyseurs sont des substances qui modifient la voie (mécanisme) d’une réaction qui à son tour augmente la vitesse d’une réaction en abaissant l’ énergie d’activation nécessaire pour que la réaction ait lieu. Un Catalyseur n’est pas détruit ou modifié au cours d’une réaction, il peut donc être réutilisé.
- Pour certaines réactions, la présence de rayonnement électromagnétique , notamment de Lumière ultraviolette , est nécessaire pour favoriser la rupture des liaisons afin de démarrer la réaction. Ceci est particulièrement vrai pour les réactions impliquant des radicaux .
Plusieurs théories permettent de calculer les vitesses de réaction au niveau moléculaire. Ce champ est appelé dynamique de réaction. La vitesse v d’une réaction de premier ordre , qui pourrait être la désintégration d’une substance A, est donnée par :
v = − d [ A ] d t = k ⋅ [ A ] . {displaystyle v=-{frac {d[{ce {A}}]}{dt}}=kcdot [{ce {A}}].} ![{displaystyle v=-{frac {d[{ce {A}}]}{dt}}=kcdot [{ce {A}}].}](https://wikimedia.org/api/rest_v1/media/math/render/svg/12291760fcaff20a02ff74abd0dfcb922664cddb)
Son intégration donne :
[ A ] ( t ) = [ A ] 0 ⋅ e − k ⋅ t . {displaystyle {ce {[A]}}(t)={ce {[A]}}_{0}cdot e^{-kcdot t}.} ![{displaystyle {ce {[A]}}(t)={ce {[A]}}_{0}cdot e^{-kcdot t}.}](https://wikimedia.org/api/rest_v1/media/math/render/svg/498c37558508e2f7297604f93bb5408dcd8c3fd4)
Ici, k est la constante de vitesse du premier ordre ayant la dimension 1/temps, [A](t) est la concentration à un instant t et [A] 0 est la concentration initiale. La vitesse d’une réaction de premier ordre ne dépend que de la concentration et des propriétés de la substance impliquée, et la réaction elle-même peut être décrite avec la demi-vie caractéristique . Plus d’une constante de temps est nécessaire pour décrire des réactions d’ordre supérieur. La dépendance à la température de la constante de vitesse suit généralement l’ équation d’Arrhenius :
k = k 0 e − E a / k B T {displaystyle k=k_{0}e^{{-E_{a}}/{k_{B}T}}} 
où E a est l’énergie d’activation et k B est la constante de Boltzmann . L’un des modèles les plus simples de vitesse de réaction est la théorie des collisions . Des modèles plus réalistes sont adaptés à un problème spécifique et incluent la théorie de l’état de transition , le calcul de la surface d’énergie potentielle , la théorie de Marcus et la théorie de Rice-Ramsperger-Kassel-Marcus (RRKM) . [20]
Types de réaction
Quatre types de base
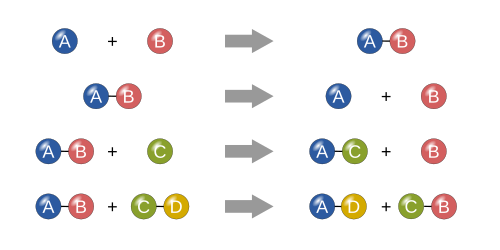 Représentation de quatre types de réactions chimiques de base : synthèse, décomposition, simple remplacement et double remplacement. Synthèse
Représentation de quatre types de réactions chimiques de base : synthèse, décomposition, simple remplacement et double remplacement. Synthèse
Dans une Réaction de synthèse, deux substances simples ou plus se combinent pour former une substance plus complexe. Ces réactions se présentent sous la forme générale :
A + B ⟶ AB {displaystyle {ce {A + B->AB}}} 
Deux ou plusieurs réactifs donnant un produit est une autre façon d’identifier une Réaction de synthèse. Un exemple de Réaction de synthèse est la combinaison de fer et de soufre pour former du sulfure de fer(II) :
8 Fe + S 8 ⟶ 8 FeS {displaystyle {ce {8Fe + S8->8FeS}}} 
Un autre exemple est l’hydrogène gazeux simple combiné avec de l’oxygène gazeux simple pour produire une substance plus complexe, telle que l’eau. [21]
Décomposition
Une Réaction de décomposition se produit lorsqu’une substance plus complexe se décompose en ses parties les plus simples. C’est donc l’opposé d’une Réaction de synthèse, et peut s’écrire [21] [22]
AB ⟶ A + B {displaystyle {ce {AB->A + B}}} 
Un exemple de Réaction de décomposition est l’ électrolyse de l’eau pour produire de l’oxygène et de l’ hydrogène gazeux :
2 H 2 O ⟶ 2 H 2 + O 2 {displaystyle {ce {2H2O->2H2 + O2}}} 
Dans une réaction de déplacement simple , un seul élément non combiné en remplace un autre dans un composé ; en d’autres termes, un élément échange sa place avec un autre élément dans un composé [21] Ces réactions se présentent sous la forme générale de :
A + BC ⟶ AC + B {displaystyle {ce {A + BC->AC + B}}} 
Un exemple d’une réaction de déplacement unique est lorsque le magnésium remplace l’hydrogène dans l’eau pour fabriquer de l’hydroxyde de magnésium et de l’hydrogène gazeux :
Mg + 2 H 2 O ⟶ Mg ( OH ) 2 + H 2 ↑ {displaystyle {ce {Mg + 2H2O->Mg(OH)2 + H2 (^)}}} 
Dans une réaction de double déplacement , les anions et les cations de deux composés changent de place et forment deux composés entièrement différents. [21] Ces réactions se présentent sous la forme générale : [22]
AB + CD ⟶ AD + CB {displaystyle {ce {AB + CD->AD + CB}}} 
Par exemple, lorsque le chlorure de baryum (BaCl 2 ) et le sulfate de magnésium (MgSO 4 ) réagissent, l’anion SO 4 2− change de place avec l’anion 2Cl − , donnant les composés BaSO 4 et MgCl 2 .
Un autre exemple de réaction de double déplacement est la réaction du nitrate de plomb (II) avec l’iodure de potassium pour former de l’iodure de plomb (II) et du nitrate de potassium :
Pb ( NO 3 ) 2 + 2 KI ⟶ PbI 2 ↓ + 2 KNO 3 {displaystyle {ce {Pb(NO3)2 + 2KI->PbI2(v) + 2KNO3}}} 
La combustion
Dans une réaction de combustion , un élément ou un composé réagit avec l’oxygène , produisant souvent de l’énergie sous forme de chaleur ou de lumière . Les réactions de combustion impliquent toujours de l’oxygène, mais impliquent aussi fréquemment un hydrocarbure .
2 C 8 H 18 ( l ) + 25 O 2 ( g ) ⟶ 16 CO 2 + 18 H 2 O ( g ) {displaystyle {ce {2C8H18(l) + 25O2(g)->16CO2 + 18H2O(g)}}} 
Une réaction de combustion peut également résulter de la réaction du carbone , du magnésium ou du soufre avec l’oxygène. [23]
2 Mg ( s ) + O 2 ⟶ 2 MgO ( s ) {displaystyle {ce {2Mg(s) + O2->2MgO(s)}}} 

Oxydation et réduction
 Illustration d’une réaction redox
Illustration d’une réaction redox  Le chlorure de sodium est formé par la réaction redox du sodium métallique et du chlore gazeux
Le chlorure de sodium est formé par la réaction redox du sodium métallique et du chlore gazeux
Les réactions redox peuvent être comprises en termes de transfert d’électrons d’une espèce impliquée ( agent réducteur ) à une autre ( agent oxydant ). Dans ce processus, la première espèce est oxydée et la seconde est réduite . Bien que suffisantes à de nombreuses fins, ces descriptions ne sont pas précisément correctes. L’Oxydation est mieux définie comme une augmentation de l’état d’Oxydation et la réduction comme une diminution de l’état d’Oxydation. En pratique, le transfert d’électrons changera toujours l’état d’Oxydation, mais de nombreuses réactions sont classées comme “redox” même si aucun transfert d’électrons ne se produit (comme celles impliquant des liaisons covalentes ). [24] [25]
Dans la réaction redox suivante, le sodium métallique dangereux réagit avec le chlore gazeux toxique pour former le composé ionique chlorure de sodium ou sel de table commun :
2 Na ( s ) + Cl 2 ( g ) ⟶ 2 NaCl ( s ) {displaystyle {ce {2Na(s) + Cl2(g)->2NaCl(s)}}} 
Dans la réaction, le sodium métallique passe d’un état d’Oxydation de 0 (car il s’agit d’un élément pur) à +1 : en d’autres termes, le sodium a perdu un électron et on dit qu’il s’est oxydé. Par contre, le chlore gazeux passe d’une Oxydation de 0 (c’est aussi un élément pur) à -1 : le chlore gagne un électron et on dit qu’il a été réduit. Parce que le chlore est celui qui est réduit, il est considéré comme l’accepteur d’électrons, ou en d’autres termes, induit l’Oxydation du sodium – ainsi le chlore gazeux est considéré comme l’agent oxydant. A l’inverse, le sodium est oxydé ou est le donneur d’électrons, et induit ainsi la réduction des autres espèces et est considéré comme l’ agent réducteur .
Lequel des réactifs impliqués serait un agent réducteur ou oxydant peut être prédit à partir de l’ électronégativité de leurs éléments. Les éléments à faible électronégativité, comme la plupart des métaux, donnent facilement des électrons et s’oxydent – ce sont des agents réducteurs. Au contraire, de nombreux ions avec des nombres d’Oxydation élevés, tels que H2O2, MnO−
4, CrO3, Cr2O2−
7, OsO4peuvent gagner un ou deux électrons supplémentaires et sont de puissants agents oxydants.
Pour certains éléments du groupe principal, le nombre d’électrons donnés ou acceptés dans une réaction redox peut être prédit à partir de la configuration électronique de l’élément Réactif. Les éléments essaient d’atteindre la configuration des gaz rares à faible énergie , et donc les métaux alcalins et les halogènes donneront et accepteront respectivement un électron. Les gaz rares eux-mêmes sont chimiquement inactifs. [26]
La réaction redox globale peut être équilibrée en combinant les demi-réactions d’Oxydation et de réduction multipliées par des coefficients tels que le nombre d’électrons perdus lors de l’Oxydation soit égal au nombre d’électrons gagnés lors de la réduction.
Une classe importante de réactions redox sont les réactions électrochimiques , où les électrons de l’alimentation électrique sont utilisés comme agent réducteur. Ces réactions sont particulièrement importantes pour la production d’éléments chimiques, comme le chlore [27] ou l’aluminium . Le processus inverse dans lequel les électrons sont libérés dans les réactions redox et peuvent être utilisés comme énergie électrique est possible et utilisé dans les batteries.
Complexation
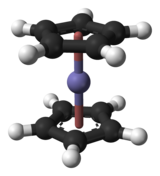 Ferrocène – un atome de fer pris en sandwich entre deux ligands C 5 H 5
Ferrocène – un atome de fer pris en sandwich entre deux ligands C 5 H 5
Dans les réactions de complexation, plusieurs ligands réagissent avec un atome métallique pour former un complexe de coordination . Ceci est réalisé en fournissant des paires isolées du ligand dans des orbitales vides de l’atome de métal et en formant des liaisons dipolaires . Les ligands sont des bases de Lewis , ils peuvent être à la fois des ions et des molécules neutres, comme le monoxyde de carbone, l’ammoniac ou l’eau. Le nombre de ligands qui réagissent avec un atome de métal central peut être trouvé en utilisant la règle des 18 électrons , en disant que les coquilles de valence d’un métal de transition accueilleront collectivement 18 électrons, alors que la symétrie du complexe résultant peut être prédite avec la théorie des champs cristallins et la théorie des champs de ligands . Les réactions de complexation comprennent également l’ échange de ligands , dans lequel un ou plusieurs ligands sont remplacés par un autre, et les processus redox qui modifient l’état d’Oxydation de l’atome métallique central. [28]
Réactions acido-basiques
Dans la théorie acide-base de Brønsted-Lowry , une réaction acide-base implique un transfert de protons (H + ) d’une espèce (l’ acide ) à une autre (la base ). Lorsqu’un proton est retiré d’un acide, l’espèce résultante est appelée base conjuguée de cet acide . Lorsque le proton est accepté par une base, l’espèce résultante est appelée acide conjugué de cette base . [29] En d’autres termes, les acides agissent comme des donneurs de protons et les bases agissent comme des accepteurs de protons selon l’équation suivante :
HA acid + B base ↽ − − ⇀ A − conjugated base + HB + conjugated acid {displaystyle {ce {{underset {acide}{HA}}+{underset {base}{B}}<=>{underset {conjuguébase}{A^{-}}}+{ underset {conjugué acide}{HB+}}}}} 
La réaction inverse est possible, et ainsi l’acide/base et la base/acide conjugués sont toujours en équilibre. L’équilibre est déterminé par les constantes de dissociation acide et basique ( K a et K b ) des substances impliquées. Un cas particulier de la réaction acide-base est la neutralisation où un acide et une base, pris exactement aux mêmes quantités, forment un sel neutre .
Les réactions acide-base peuvent avoir des définitions différentes selon le concept acide-base utilisé. Certains des plus courants sont :
- Définition d’ Arrhenius : Les acides se dissocient dans l’eau en libérant des ions H 3 O + ; les bases se dissocient dans l’eau en libérant des ions OH − .
- Définition de Brønsted–Lowry : Les acides sont des donneurs de protons (H + ), les bases sont des accepteurs de protons ; cela inclut la définition d’Arrhenius.
- Définition de Lewis : les acides sont des accepteurs de paires d’électrons, les bases sont des donneurs de paires d’électrons ; cela inclut la définition de Brønsted-Lowry.
Précipitation
 Précipitation
Précipitation
La précipitation est la formation d’un solide dans une solution ou à l’intérieur d’un autre solide lors d’une réaction Chimique. Cela se produit généralement lorsque la concentration des ions dissous dépasse la limite de solubilité [30] et forme un sel insoluble. Ce processus peut être facilité par l’ajout d’un agent précipitant ou par l’élimination du solvant. Une précipitation rapide donne un résidu amorphe ou microcristallin et un processus lent peut produire des monocristaux . Ce dernier peut également être obtenu par recristallisation dans des sels microcristallins. [31]
Réactions à l’état solide
Des réactions peuvent avoir lieu entre deux solides. Cependant, en raison des taux de diffusion relativement faibles dans les solides, les réactions chimiques correspondantes sont très lentes par rapport aux réactions en phase liquide et gazeuse. Ils sont accélérés en augmentant la température de réaction et en divisant finement le Réactif pour augmenter la surface de contact. [32]
Réactions à l’interface solide|gaz
La réaction peut avoir lieu à l’interface solide|gaz, sur des surfaces à très basse pression comme l’ ultra-vide . Grâce à la microscopie à effet tunnel , il est possible d’observer des réactions à l’interface solide|gaz dans l’espace réel, si l’échelle de temps de la réaction est dans la bonne plage. [33] [34] Les réactions à l’interface solide|gaz sont dans certains cas liées à la catalyse.
Réactions photochimiques
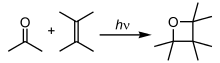 Dans cette réaction de Paterno-Büchi , un groupe carbonyle photoexcité est ajouté à une oléfine non excitée , donnant un oxétane .
Dans cette réaction de Paterno-Büchi , un groupe carbonyle photoexcité est ajouté à une oléfine non excitée , donnant un oxétane .
Dans les réactions photochimiques , les atomes et les molécules absorbent l’énergie ( photons ) de la lumière d’éclairage et se transforment en un état excité . Ils peuvent ensuite libérer cette énergie en cassant des liaisons chimiques, produisant ainsi des radicaux. Les réactions photochimiques comprennent les réactions hydrogène-oxygène, la polymérisation radicalaire , les réactions en chaîne et les réactions de réarrangement . [35]
De nombreux processus importants impliquent la photochimie. Le premier exemple est la photosynthèse , dans laquelle la plupart des plantes utilisent l’énergie solaire pour convertir le dioxyde de carbone et l’eau en glucose , éliminant l’oxygène comme sous-produit. Les humains dépendent de la photochimie pour la formation de la vitamine D, et la vision est initiée par une réaction photochimique de la rhodopsine . [15] Chez les lucioles , une enzyme dans l’abdomen catalyse une réaction qui se traduit par la bioluminescence . [36] De nombreuses réactions photochimiques importantes, telles que la formation d’ozone, se produisent dans l’atmosphère terrestre et constituentchimie atmosphérique .
Catalyse
 Diagramme d’énergie potentielle schématique montrant l’effet d’un Catalyseur dans une réaction Chimique Endothermique. La présence d’un Catalyseur ouvre une voie de réaction différente (en rouge) avec une énergie d’activation plus faible. Le résultat final et la thermodynamique globale sont les mêmes.
Diagramme d’énergie potentielle schématique montrant l’effet d’un Catalyseur dans une réaction Chimique Endothermique. La présence d’un Catalyseur ouvre une voie de réaction différente (en rouge) avec une énergie d’activation plus faible. Le résultat final et la thermodynamique globale sont les mêmes.  Les catalyseurs hétérogènes solides sont plaqués sur des mailles dans des convertisseurs catalytiques en céramique afin de maximiser leur surface. Ce convertisseur d’échappement provient d’une Peugeot 106 S2 1100
Les catalyseurs hétérogènes solides sont plaqués sur des mailles dans des convertisseurs catalytiques en céramique afin de maximiser leur surface. Ce convertisseur d’échappement provient d’une Peugeot 106 S2 1100
En catalyse , la réaction ne se déroule pas directement, mais par réaction avec une troisième substance appelée Catalyseur . Bien que le Catalyseur participe à la réaction, il est ramené à son état d’origine à la fin de la réaction et n’est donc pas consommé. Cependant, il peut être inhibé, désactivé ou détruit par des processus secondaires. Les catalyseurs peuvent être utilisés dans une phase différente ( hétérogène ) ou dans la même phase ( homogène ) que les réactifs. Dans la catalyse hétérogène, les processus secondaires typiques incluent la cokéfaction où le Catalyseur est recouvert de polymèreproduits secondaires. De plus, les catalyseurs hétérogènes peuvent se dissoudre dans la solution dans un système solide-liquide ou s’évaporer dans un système solide-gaz. Les catalyseurs ne peuvent qu’accélérer la réaction – les produits chimiques qui ralentissent la réaction sont appelés inhibiteurs. [37] [38] Les substances qui augmentent l’activité des catalyseurs sont appelées promoteurs, et les substances qui désactivent les catalyseurs sont appelées poisons catalytiques. Avec un Catalyseur, une réaction cinétiquement inhibée par une énergie d’activation élevée peut avoir lieu en contournant cette énergie d’activation.
Les catalyseurs hétérogènes sont généralement des solides, réduits en poudre afin de maximiser leur surface. Les métaux du groupe du platine et d’autres métaux de transition, qui sont utilisés dans les hydrogénations , le reformage catalytique et dans la synthèse de produits chimiques de base tels que l’acide nitrique et l’ammoniac , revêtent une importance particulière dans la catalyse hétérogène . Les acides sont un exemple de Catalyseur homogène, ils augmentent la nucléophilie des carbonyles, permettant une réaction qui ne se déroulerait pas autrement avec des électrophiles. L’avantage des catalyseurs homogènes est la facilité de leur mélange avec les réactifs, mais ils peuvent également être difficiles à séparer des produits. Par conséquent, les catalyseurs hétérogènes sont préférés dans de nombreux procédés industriels. [39]
Réactions en chimie organique
En chimie organique, en plus des réactions d’Oxydation, de réduction ou acido-basique, un certain nombre d’autres réactions peuvent avoir lieu qui impliquent des liaisons covalentes entre des atomes de carbone ou du carbone et des hétéroatomes (tels que l’oxygène, l’azote, les halogènes , etc.). De nombreuses réactions spécifiques en chimie organique portent le nom de réactions désignées d’après leurs découvreurs.
Substitution
Dans une réaction de substitution , un groupe fonctionnel dans un composé Chimique particulier est remplacé par un autre groupe. [40] Ces réactions peuvent être distinguées par le type d’espèce de substitution dans une substitution nucléophile , électrophile ou radicale .
 Mécanisme S N 1
Mécanisme S N 1 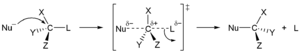 Mécanisme S N 2
Mécanisme S N 2
Dans le premier type, un nucléophile , un atome ou une molécule avec un excès d’électrons et donc une charge négative ou une charge partielle , remplace un autre atome ou une partie de la molécule “substrat”. La paire d’électrons du nucléophile attaque le substrat en formant une nouvelle liaison, tandis que le groupe partant part avec une paire d’électrons. Le nucléophile peut être électriquement neutre ou chargé négativement, tandis que le substrat est typiquement neutre ou chargé positivement. Des exemples de nucléophiles sont l’ ion hydroxyde , les alcoxydes , les amines et les halogénures . Ce type de réaction se retrouve principalement dans les hydrocarbures aliphatiques , et rarement danshydrocarbure aromatique . Ces derniers ont une densité électronique élevée et n’entrent en substitution aromatique nucléophile qu’avec des groupes électroattracteurs très puissants . La substitution nucléophile peut avoir lieu par deux mécanismes différents, S N 1 et S N 2 . Dans leurs noms, S signifie substitution, N pour nucléophile, et le nombre représente l’ ordre cinétique de la réaction, unimoléculaire ou bimoléculaire. [41]
 Les trois étapes d’une réaction S N 2 . Le nucléophile est vert et le groupe partant est rouge
Les trois étapes d’une réaction S N 2 . Le nucléophile est vert et le groupe partant est rouge  La réaction S N 2 provoque une inversion stéréo (inversion de Walden)
La réaction S N 2 provoque une inversion stéréo (inversion de Walden)
La réaction S N 1 se déroule en deux étapes. Tout d’abord, le groupe partant est éliminé en créant un carbocation . Ceci est suivi d’une réaction rapide avec le nucléophile. [42]
Dans le mécanisme S N 2, le nucléophile forme un état de transition avec la molécule attaquée, et alors seulement le groupe partant est clivé. Ces deux mécanismes diffèrent par la stéréochimie des produits. S N 1 conduit à l’addition non stéréospécifique et n’aboutit pas à un centre chiral, mais plutôt à un ensemble d’ isomères géométriques ( cis/trans ). En revanche, une inversion ( inversion de Walden ) de la stéréochimie préexistante est observée dans le mécanisme S N 2 . [43]
La substitution électrophile est la contrepartie de la substitution nucléophile en ce que l’atome ou la molécule attaquant, un électrophile , a une faible densité électronique et donc une charge positive. Les électrophiles typiques sont l’atome de carbone des groupes carbonyle , les carbocations ou les cations soufre ou nitronium . Cette réaction a lieu presque exclusivement dans les hydrocarbures aromatiques, où elle est appelée substitution aromatique électrophile .. L’attaque électrophile entraîne le soi-disant complexe σ, un état de transition dans lequel le système aromatique est aboli. Ensuite, le groupe partant, généralement un proton, est séparé et l’aromaticité est restaurée. Une alternative à la substitution aromatique est la substitution aliphatique électrophile. Il est similaire à la substitution aliphatique nucléophile et a également deux types principaux, S E 1 et S E 2 [44]
 Mécanisme de substitution aromatique électrophile
Mécanisme de substitution aromatique électrophile
Dans le troisième type de réaction de substitution, la substitution radicalaire, la particule attaquante est un radical . [40] Ce processus prend généralement la forme d’une réaction en chaîne , par exemple dans la réaction d’alcanes avec des halogènes. Dans la première étape, la lumière ou la chaleur désintègre les molécules contenant des halogènes produisant les radicaux. Ensuite, la réaction se déroule comme une avalanche jusqu’à ce que deux radicaux se rencontrent et se recombinent. [45]
X ⋅ + R − H ⟶ X − H + R ⋅ {displaystyle {ce {X. + RH -> XH + R.}}} 

Addition et élimination
L’ addition et sa contrepartie, l’ élimination , sont des réactions qui modifient le nombre de substituants sur l’atome de carbone, et forment ou clivent des liaisons multiples . Des liaisons doubles et triples peuvent être produites en éliminant un groupe partant approprié. Semblable à la substitution nucléophile, il existe plusieurs mécanismes de réaction possibles qui sont nommés d’après l’ordre de réaction respectif. Dans le mécanisme E1, le groupe partant est éjecté en premier, formant un carbocation. L’étape suivante, la formation de la double liaison, a lieu avec élimination d’un proton ( déprotonation). L’ordre de sortie est inversé dans le mécanisme E1cb, c’est-à-dire que le proton est séparé en premier. Ce mécanisme nécessite la participation d’une base. [46] En raison des conditions similaires, les deux réactions dans l’élimination E1 ou E1cb sont toujours en concurrence avec la substitution S N 1. [47]
 Élimination E1
Élimination E1  Élimination E1cb
Élimination E1cb  Élimination E2
Élimination E2
Le mécanisme E2 nécessite également une base, mais là l’attaque de la base et l’élimination du groupe partant se déroulent simultanément et ne produisent aucun intermédiaire ionique. Contrairement aux éliminations E1, différentes configurations stéréochimiques sont possibles pour le produit de réaction dans le mécanisme E2, car l’attaque de la base se produit préférentiellement en position anti par rapport au groupe partant. En raison des conditions et des réactifs similaires, l’élimination de E2 est toujours en compétition avec la substitution S N 2 . [48]
![]()
![]() Addition électrophile de bromure d’hydrogène
Addition électrophile de bromure d’hydrogène
La contrepartie de l’élimination est l’addition où les doubles ou triples liaisons sont converties en liaisons simples. Semblables aux réactions de substitution, il existe plusieurs types d’additions qui se distinguent par le type de particule attaquante. Par exemple, dans l’ addition électrophile de bromure d’hydrogène, un électrophile (proton) attaque la double liaison formant un carbocation , qui réagit alors avec le nucléophile (brome). Le carbocation peut être formé de chaque côté de la double liaison en fonction des groupes attachés à ses extrémités, et la configuration préférée peut être prédite avec la règle de Markovnikov . [49]Cette règle stipule que “Dans l’addition hétérolytique d’une molécule polaire à un alcène ou un alcyne, l’atome (ou la partie) le plus électronégatif (nucléophile) de la molécule polaire s’attache à l’atome de carbone portant le plus petit nombre d’atomes d’hydrogène.” [50]
Si l’addition d’un groupe fonctionnel a lieu au niveau de l’atome de carbone le moins substitué de la double liaison, alors la substitution électrophile avec des acides n’est pas possible. Dans ce cas, il faut utiliser la réaction d’hydroboration-Oxydation , où dans la première étape, l’ atome de bore agit comme électrophile et s’ajoute à l’atome de carbone le moins substitué. Lors de la deuxième étape, l’ hydroperoxyde nucléophile ou l’ anion halogène attaque l’atome de bore. [51]
Alors que l’addition aux alcènes et alcynes riches en électrons est principalement électrophile, l’ addition nucléophile joue un rôle important pour les liaisons multiples carbone-hétéroatome, et en particulier son représentant le plus important, le groupe carbonyle. Ce processus est souvent associé à une élimination, de sorte qu’après la réaction le groupe carbonyle est à nouveau présent. Elle est donc appelée réaction d’addition-élimination et peut se produire dans les dérivés d’acide carboxylique tels que les chlorures, les esters ou les anhydrides. Cette réaction est souvent catalysée par des acides ou des bases, où les acides augmentent par l’électrophilie du groupe carbonyle en se liant à l’atome d’oxygène, tandis que les bases renforcent la nucléophilie du nucléophile attaquant. [52]
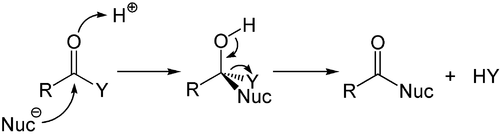 Mécanisme d’addition-élimination catalysé par l’acide
Mécanisme d’addition-élimination catalysé par l’acide
L’addition nucléophile d’un carbanion ou d’un autre nucléophile à la double liaison d’un composé carbonyle alpha, bêta insaturé peut se dérouler via la réaction de Michael , qui appartient à la classe plus large des additions conjuguées . C’est l’une des méthodes les plus utiles pour la formation douce de liaisons C – C. [53] [54] [55]
Certaines additions qui ne peuvent pas être exécutées avec des nucléophiles et des électrophiles, peuvent être réussies avec des radicaux libres. Comme pour la substitution radicalaire, l’ addition radicalaire se déroule comme une réaction en chaîne, et ces réactions sont à la base de la polymérisation radicalaire . [56]
Autres mécanismes de réaction organique
![]()
![]() Le réarrangement Cope du 3-méthyl-1,5-hexadiène
Le réarrangement Cope du 3-méthyl-1,5-hexadiène  Mécanisme d’une réaction de Diels-Alder
Mécanisme d’une réaction de Diels-Alder  Chevauchement orbital dans une réaction de Diels-Alder
Chevauchement orbital dans une réaction de Diels-Alder
Dans une réaction de réarrangement , le squelette carboné d’une molécule est réarrangé pour donner un isomère structurel de la molécule d’origine. Celles-ci incluent des réactions de déplacement d’hydrure telles que le réarrangement de Wagner-Meerwein , où un groupe hydrogène , alkyle ou aryle migre d’un carbone vers un carbone voisin. La plupart des réarrangements sont associés à la rupture et à la formation de nouvelles liaisons carbone-carbone. D’autres exemples sont la réaction sigmatropique telle que le réarrangement de Cope . [57]
Les réarrangements cycliques comprennent les cycloadditions et, plus généralement, les réactions péricycliques , dans lesquelles deux ou plusieurs molécules contenant une double liaison forment une molécule cyclique. Un exemple important de réaction de cycloaddition est la réaction de Diels – Alder (la soi-disant [4 + 2] cycloaddition) entre un diène conjugué et un alcène substitué pour former un système cyclohexène substitué . [58]
Le fait qu’une certaine cycloaddition se déroule dépend des orbitales électroniques des espèces participantes, car seules les orbitales avec le même signe de fonction d’onde se chevaucheront et interagiront de manière constructive pour former de nouvelles liaisons. La cycloaddition est généralement assistée par la lumière ou la chaleur. Ces perturbations se traduisent par une disposition différente des électrons dans l’état excité des molécules impliquées et donc par des effets différents. Par exemple, les réactions [4+2] Diels-Alder peuvent être assistées par la chaleur alors que la cycloaddition [2+2] est induite sélectivement par la lumière. [59] En raison du caractère orbital, le potentiel de développement de produits stéréoisomères lors de la cycloaddition est limité, comme décrit par les règles de Woodward-Hoffmann . [60]
Réactions biochimiques
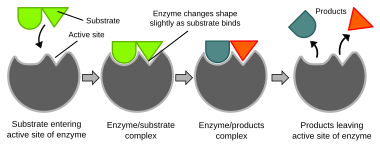 Illustration du modèle d’ajustement induit de l’activité enzymatique
Illustration du modèle d’ajustement induit de l’activité enzymatique
Les réactions biochimiques sont principalement contrôlées par des enzymes . Ces protéines peuvent catalyser spécifiquement une seule réaction, de sorte que les réactions peuvent être contrôlées très précisément. La réaction a lieu dans le site actif , une petite partie de l’enzyme qui se trouve généralement dans une fente ou une poche tapissée de résidus d’ acides aminés, et le reste de l’enzyme est principalement utilisé pour la stabilisation. L’action catalytique des enzymes repose sur plusieurs mécanismes dont la forme moléculaire (« ajustement induit »), la contrainte de liaison, la proximité et l’orientation des molécules par rapport à l’enzyme, le don ou le retrait de protons (catalyse acide/base), les interactions électrostatiques et bien d’autres. [61]
Les réactions biochimiques qui se produisent dans les organismes vivants sont collectivement connues sous le nom de métabolisme . Parmi ses mécanismes les plus importants figure l’ anabolisme , dans lequel différents processus contrôlés par l’ ADN et les enzymes entraînent la production de grosses molécules telles que des protéines et des glucides à partir d’unités plus petites. [62] La bioénergétique étudie les sources d’énergie pour de telles réactions. Une source d’énergie importante est le glucose , qui peut être produit par les plantes via la photosynthèse ou assimilé à partir des aliments. Tous les organismes utilisent cette énergie pour produire de l’ adénosine triphosphate(ATP), qui peut ensuite être utilisé pour dynamiser d’autres réactions.
Applications
 Réaction aluminothermique se produisant dans le soudage ferroviaire. Peu de temps après, la fonte liquide s’écoule dans le moule autour de l’espace du rail
Réaction aluminothermique se produisant dans le soudage ferroviaire. Peu de temps après, la fonte liquide s’écoule dans le moule autour de l’espace du rail
Les réactions chimiques sont au cœur du génie Chimique où elles sont utilisées pour la synthèse de nouveaux composés à partir de matières premières naturelles telles que le pétrole et les minerais . Il est essentiel de rendre la réaction aussi efficace que possible, en maximisant le rendement et en minimisant la quantité de réactifs, les apports d’énergie et les déchets. Les catalyseurs sont particulièrement utiles pour réduire l’énergie nécessaire à la réaction et augmenter sa vitesse de réaction . [63] [64]
Certaines réactions spécifiques ont leurs applications de niche. Par exemple, la réaction aluminothermique est utilisée pour générer de la lumière et de la chaleur dans la pyrotechnie et le soudage . Bien qu’il soit moins contrôlable que le soudage oxy-combustible , le soudage à l’ arc et le soudage par étincelage plus conventionnels , il nécessite beaucoup moins d’équipement et est toujours utilisé pour réparer les rails, en particulier dans les régions éloignées. [65]
Surveillance
Les mécanismes de surveillance des réactions chimiques dépendent fortement de la vitesse de réaction. Des processus relativement lents peuvent être analysés in situ pour les concentrations et les identités des ingrédients individuels. Les outils importants de l’analyse en temps réel sont la mesure du pH et l’analyse des spectres d’absorption optique (couleur) et d’émission. Une méthode moins accessible mais plutôt efficace consiste à introduire un isotope Radioactif dans la réaction et à surveiller son évolution dans le temps et sa destination ; cette méthode est souvent utilisée pour analyser la redistribution des substances dans le corps humain. Les réactions plus rapides sont généralement étudiées avec la spectroscopie laser ultrarapide où l’utilisation de lasers femtosecondes permet de surveiller les états de transition de courte durée à un temps réduit à quelques femtosecondes. [66]
Voir également
-
![]()
![]() Portail de la chimie
Portail de la chimie
| Wikiquote a des citations liées à la réaction Chimique . |
- Équation Chimique
- Réaction Chimique
- Substrat
- Réactif
- Catalyseur
- Produit
- Modèle de réaction Chimique
- Chimiste
- Chimie
- La combustion
- Réactif limitant
- Liste des réactions organiques
- Équilibre de la masse
- Réversibilité microscopique
- Réaction organique
- Analyse cinétique de l’avancement de la réaction
- Réaction réversible
Références
- ^ IUPAC , Compendium de terminologie Chimique , 2e éd. (le “Livre d’or”) (1997). Version corrigée en ligne : (2006–) « réaction Chimique ». doi : 10.1351/goldbook.C01033
- ^ Weyer, J. (1973). “Neuere Interprétationsmöglichkeiten der Alchemie”. Chemie in unserer Zeit . 7 (6): 177–181. doi : 10.1002/ciuz.19730070604 .
- ^ Voir Newman, William R. (2004). Promethean Ambitions: Alchemy and the Quest to Perfect Nature . Chicago : presse de l’université de Chicago. ISBN 9780226575247.
- ^ Kraus, Paul (1942-1943). Jâbir ibn Hayyân : Contribution à l’histoire des idées scientifiques dans l’Islam. I. Le corpus des écrits jâbiriens. II. Jâbir et la science grecque . Le Caire : Institut Français d’Archéologie Orientale . ISBN 9783487091150. OCLC 468740510 ., vol. II, p. 41–42.
- ^ Karpenko, Vladimir; En ligneNorris, John A. (2002). “Le vitriol dans l’histoire de la chimie” . Chemické listy . 96 (12): 997-1005.
- ^ Friedman, Leonard J.; Friedman, Samantha J. (2008). L’histoire du processus d’acide sulfurique de contact (PDF) . Boca Raton, Floride : Acid Engineering & Consulting, Inc.
- ^ Étranges, Anthony N. (2000). “L’industrie allemande des carburants synthétiques, 1935–1940”. Dans Lesch, John E. (éd.). L’industrie Chimique allemande au XXe siècle . Éditeurs académiques Kluwer . p. 170. ISBN 978-0-7923-6487-0.
- ^ Broc, pp. 34–55
- ^ Brock , pp. 104–107
- ^ Myers, Richard (2009). Les bases de la chimie . Groupe d’édition Greenwood . p. 55. ISBN 978-0-313-31664-7.
- ^ IUPAC , Compendium de terminologie Chimique , 2e éd. (le “Livre d’or”) (1997). Version corrigée en ligne : (2006–) « équation de réaction Chimique ». doi : 10.1351/goldbook.C01034
- ^ Corey, EJ (1988). “Conférence Robert Robinson. Pensée rétrosynthétique? Essentiels et exemples”. Examens de la société Chimique . 17 : 111–133. doi : 10.1039/CS9881700111 .
- ^ IUPAC , Compendium de terminologie Chimique , 2e éd. (le “Livre d’or”) (1997). Version corrigée en ligne : (2006–) « réaction élémentaire ». doi : 10.1351/goldbook.E02035
- ^ Frenking, Gernot (2006). “Elementarreaktionen”. Römpp Chemie-Lexikon . Thième .
- ^ un b Kandori, Hideki (2006). “Protéines de liaison rétiniennes”. Dans Dugave, Christophe (dir.). Isomérisation cis-trans en biochimie . Wiley-VCH . p. 56. ISBN 978-3-527-31304-4.
- ^ Atkins , p. 114.
- ^ Atkins , pp. 106-108
- ^ un b Web de réaction
- ^ Atkins , p. 150
- ^ Atkins , p. 963
- ^ a b c d Réagir ou ne pas réagir ? Archivé le 10/01/2015 au Wayback Machine Utah State Office of Education. Récupéré le 4 juin 2011.
- ^ un b Les six types de réaction – La Cavalcade de la Chimie. Récupéré le 11 février 2016
- ^ Wilbraham, Matta, Waterman, Stanley, Antony, Michael, Edward, Dennis (2012). Chimie . Person. pages 734–735. ISBN 978-0-13-322662-1.{{cite book}}: CS1 maint: multiple names: authors list (link)
- ^ Glusker, Jenny P. (1991). “Aspects structurels de la ligature métallique aux groupes fonctionnels dans les protéines”. Dans Christian B. Anfinsen (éd.). Avancées en chimie des protéines . Vol. 42. San Diego : presse académique . p. 7. ISBN 978-0-12-034242-6.
- ^ Guo, Liang-Hong; Allen, H.; En ligneHill, O. (1991). “Électrochimie directe des protéines et des enzymes”. Dans AG Sykes (éd.). Progrès en chimie inorganique . Vol. 36. San Diego : presse académique . p. 359.ISBN _ 978-0-12-023636-7.
- ^ Wiberg , pp. 289–290
- ^ Wiberg , p. 409
- ^ Wiberg , pp. 1180-1205
- ^ IUPAC , Compendium de terminologie Chimique , 2e éd. (le “Livre d’or”) (1997). Version corrigée en ligne : (2006–) ” couple acide-base conjugué “. doi : 10.1351/goldbook.C01266
- ^ IUPAC , Compendium de terminologie Chimique , 2e éd. (le “Livre d’or”) (1997). Version corrigée en ligne : (2006–) « précipitations ». doi : 10.1351/goldbook.P04795
- ^ Wingender, Jörg; Ortanderl, Stefanie (juillet 2009). “Ausfällung”. Römpp Chemie-Lexikon . Thième .
- ^ Meyer, H. Jürgen (2007). “Festkörperchemie”. Dans Erwin Riedel (éd.). Modern Inorganic Chemistry (en allemand) (3e éd.). de Gruyter . p. 171. ISBN 978-3-11-019060-1.
- ^ Winterlin, J. (1997). “Taux de réaction atomique et macroscopique d’une réaction catalysée en surface”. Sciences . 278 (5345): 1931–4. Bibcode : 1997Sci…278.1931W . doi : 10.1126/science.278.5345.1931 . PMID 9395392 .
- ^ Waldmann, T.; Künzel, D.; Hôte, HE ; Gross, A.; Behm, RJR (2012). “L’Oxydation d’un Adlayer organique : Une vue d’ensemble”. Journal de l’American Chemical Society . 134 (21): 8817–8822. doi : 10.1021/ja302593v . PMID 22571820 .
- ^ Atkins , pp. 937–950
- ^ Saunders, David Stanley (2002). Horloges d’insectes (troisième éd.). Amsterdam : Elsevier . p. 179. ISBN 978-0-444-50407-4.
- ^ IUPAC , Compendium de terminologie Chimique , 2e éd. (le “Livre d’or”) (1997). Version corrigée en ligne : (2006–) « Catalyseur ». doi : 10.1351/goldbook.C00876
- ^ IUPAC , Compendium de terminologie Chimique , 2e éd. (le “Livre d’or”) (1997). Version corrigée en ligne : (2006–) ” inhibiteur “. doi : 10.1351/goldbook.I03035
- ^ Elschenbroich, Christoph (2008). Organometallchemie (6e éd.). Wiesbaden : Vieweg+Teubner Verlag . p. 263.ISBN _ 978-3-8351-0167-8.
- ^ a b March, Jerry (1985), Chimie organique avancée: réactions, mécanismes et structure (3e éd.), New York: Wiley, ISBN 0-471-85472-7
- ^ Hartshorn, SR (1973). Substitution Nucléophile Aliphatique . Londres : Cambridge University Press . p. 1. ISBN 978-0-521-09801-4.
- ^ Bateman, Leslie C.; Église, Mervyn G.; Hughes, Edward D.; Ingold, Christopher K.; Taher, Nazeer Ahmed (1940). “188. Mécanisme de substitution à un atome de carbone saturé. Partie XXIII. Une démonstration cinétique de la solvolyse unimoléculaire des halogénures d’alkyle. (Section E) une discussion générale”. Journal of the Chemical Society : 979. doi : 10.1039/JR9400000979 .
- ^ Brückner , pp. 63–77
- ^ Brückner , pp. 203–206
- ^ Brückner , p. 16
- ^ Brückner , p. 192
- ^ Brückner , p. 183
- ^ Brückner , p. 172
- ^ Wiberg , pp. 950, 1602
- ^ IUPAC , Compendium de terminologie Chimique , 2e éd. (le “Livre d’or”) (1997). Version corrigée en ligne : (2006–) ” Règle de Markownikoff “. doi : 10.1351/goldbook.M03707
- ^ Brückner , p. 125
- ^ Latscha, Hans Peter; Kazmaier, Uli; Klein, Helmut Alfons (2008). Organische Chemie : Chemie-basiswissen II (en allemand). Vol. 2 (6e éd.). Springer . p. 273.ISBN _ 978-3-540-77106-7.
- ^ Réactions organiques . 2004. doi : 10.1002/0471264180 . ISBN 978-0-471-26418-7.
- ^ Chasse, Ian. “Chapitre 18: Enols et Enolates – La réaction de Michael Addition” . Université de Calgary.
- ^ Brückner , p. 580
- ^ Lechner, Manfred; Gehrke, Klaus; En ligneNordmeier, Eckhard (2003). Chimie macromoléculaire (3e éd.). Bâle : Birkhäuser . p. 53–65. ISBN 978-3-7643-6952-1.
- ^ Renard, Marye Anne; En ligneWhitesell, James K. (2004). Chimie organique (troisième éd.). Jones et Bartlett . p. 699.ISBN _ 978-0-7637-2197-8.
- ^ Diels, O.; Alder, K. (1928). “Synthesen in der hydroaromatischen Reihe”. Annalen der Chemie de Justus Liebig . 460 : 98–122. doi : 10.1002/jlac.19284600106 .
- ^ Bruckner, pp. 637–647
- ^ Woodward, RB; En ligneHoffmann, R. (1965). “Stéréochimie des réactions électrocycliques”. Journal de l’American Chemical Society . 87 (2): 395–397. doi : 10.1021/ja01080a054 .
- ^ Karlson, Peter; Doenecke, Detlef; Koolman, janvier; Fuchs, Georg ; Gerok, Wolfgang (2005). Karlson Biochemistry and Pathobiochemistry (en allemand) (16e éd.). Thième . p. 55–56. ISBN 978-3-13-357815-8.
- ^ IUPAC , Compendium de terminologie Chimique , 2e éd. (le “Livre d’or”) (1997). Version corrigée en ligne : (2006–) ” anabolisme “. doi : 10.1351/goldbook.A00314
- ^ Emig, Gerhard; Klemm, Elias (2005). Chimie technique (en allemand) (5e éd.). Springer . p. 33–34. ISBN 978-3-540-23452-4.
- ^ Trost, B. (1991). “L’économie atomique – une recherche d’efficacité synthétique”. Sciences . 254 (5037): 1471–1477. Bib code : 1991Sci …254.1471T . doi : 10.1126/science.1962206 . PMID 1962206 .
- ^ Weismantel, Guy E (1999). John J. McKetta (éd.). Encyclopédie du traitement Chimique et de la conception . Vol. 67. CRC Appuyez sur . p. 109. ISBN 978-0-8247-2618-8 https://books.google.com/books?id=MfjDlUe8Kc0C&pg=PA109 . {{cite encyclopedia}}: Manquant ou vide |title=( aide )
- ^ Atkins , p. 987
Bibliographie
- Atkins, Peter W.; Julio de Paula (2006). Chimie physique (4e éd.). Weinheim : Wiley-VCH . ISBN 978-3-527-31546-8.
- En ligneBrock, William H. (1997). Viewegs Geschichte der Chemie (en allemand). Braunschweig : Vieweg . ISBN 978-3-540-67033-9.
- Bruckner, Reinhard (2004). Reaktionsmechanismen (en allemand) (3e éd.). Munich : Spektrum Akademischer Verlag. ISBN 978-3-8274-1579-0.
- Wiberg, Egon, Wiberg, Nils et Holleman, Arnold Frederick (2001). Chimie inorganique . Presse Académique . ISBN 978-0-12-352651-9.{{cite book}}: CS1 maint: multiple names: authors list (link)
- « Action Chimique » . Encyclopædia Britannica . Vol. 6 (11e éd.). 1911. pp. 26–33.