Fibrose kystique
La fibrose kystique ( FK ) est une maladie génétique qui affecte principalement les poumons , mais aussi le pancréas , le foie , les reins et l’intestin . [1] [5] Les problèmes à long terme incluent la difficulté à respirer et à cracher du mucus à la suite d’ infections pulmonaires fréquentes . [1] D’autres signes et symptômes peuvent inclure des infections des sinus , une croissance médiocre , des selles grasses , un hippocratisme digital des doigts et des orteils etl’infertilité chez la plupart des hommes. [1] Différentes personnes peuvent présenter différents degrés de symptômes. [1]
| Fibrose kystique | |
|---|---|
| Autres noms | Mucoviscidose |
 |
|
| Spécialité | Génétique médicale , pneumologie |
| Les symptômes | Difficulté à respirer , crachats de mucus , croissance ralentie , selles grasses [1] |
| Début habituel | Symptômes reconnaissables ~ 6 mois [2] |
| Durée | À vie [3] |
| causes | Génétique ( autosomique récessif ) [1] |
| Méthode diagnostique | Test de la sueur , tests génétiques [1] |
| Traitement | Antibiotiques , remplacement des enzymes pancréatiques , transplantation pulmonaire [1] |
| Pronostic | Espérance de vie entre 42 et 50 ans (monde développé) [4] |
| La fréquence | 1 sur 3 000 ( d’Europe du Nord ) [1] |
La mucoviscidose est transmise de manière autosomique récessive . [1] Elle est causée par la présence de mutations dans les deux copies du gène de la protéine régulatrice de la conductance transmembranaire de la fibrose kystique (CFTR). [1] Ceux qui ont une seule copie de travail sont porteurs et généralement en bonne santé. [3] Le CFTR est impliqué dans la production de sueur, de fluides digestifs et de mucus. [6] Lorsque le CFTR n’est pas fonctionnel, les sécrétions qui sont généralement minces deviennent plutôt épaisses. [7] La condition est diagnostiquée par un test de la sueur et des tests génétiques . [1]Le dépistage des nourrissons à la naissance a lieu dans certaines régions du monde. [1]
Il n’existe aucun remède connu pour la fibrose kystique. [3] Les infections pulmonaires sont traitées avec des antibiotiques qui peuvent être administrés par voie intraveineuse, inhalée ou orale. [1] Parfois, l’antibiotique azithromycine est utilisé à long terme. [1] Une solution saline hypertonique inhalée et du salbutamol peuvent également être utiles. [1] La transplantation pulmonaire peut être une option si la fonction pulmonaire continue de se détériorer. [1] Le remplacement des enzymes pancréatiques et la supplémentation en vitamines liposolubles sont importants, en particulier chez les jeunes. [1] Techniques de dégagement des voies respiratoires telles quela physiothérapie thoracique présente certains avantages à court terme, mais les effets à long terme ne sont pas clairs. [8] L’espérance de vie moyenne se situe entre 42 et 50 ans dans le monde développé . [4] [9] Les problèmes pulmonaires sont responsables du décès de 80 % des personnes atteintes de mucoviscidose. [1]
La mucoviscidose est plus fréquente chez les personnes d’ ascendance nord-européenne et affecte environ un nouveau-né sur 3 000. [1] Environ une personne sur 25 est porteuse. [3] Il est le moins courant chez les Africains et les Asiatiques. [1] Elle a été reconnue pour la première fois comme une maladie spécifique par Dorothy Andersen en 1938, avec des descriptions qui correspondent à la condition remontant au moins aussi loin que 1595. [5] Le nom “fibrose kystique” fait référence à la fibrose caractéristique et aux kystes qui se forment au sein du pancréas . [5] [10]
 Lire des médias Résumé vidéo ( script )
Lire des médias Résumé vidéo ( script )
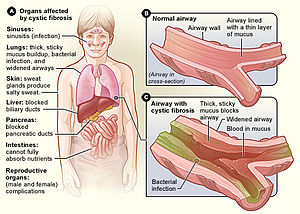
Signes et symptômes
 Problèmes de santé associés à la fibrose kystique
Problèmes de santé associés à la fibrose kystique
La fibrose kystique se manifeste généralement tôt dans la vie. Les nouveau-nés et les nourrissons atteints de mucoviscidose ont tendance à avoir des selles fréquentes, volumineuses et graisseuses (résultant d’ une malabsorption ) et ont un poids insuffisant pour leur âge . [11] 15 à 20 % des nouveau-nés ont leur intestin grêle bloqué par du méconium , nécessitant souvent une intervention chirurgicale pour corriger. [11] Les nouveau-nés ont parfois une jaunisse néonatale due au blocage des voies biliaires . [11] Les enfants atteints de fibrose kystique perdent trop de sel dans leur sueur et les parents remarquent souvent que le sel se cristallise sur la peau ou un goût salé lorsqu’ils embrassent leur enfant. [11]
La principale cause de morbidité et de décès chez les personnes atteintes de mucoviscidose est une maladie pulmonaire progressive, qui finit par entraîner une insuffisance respiratoire . [12] Cela commence généralement par une infection respiratoire prolongée qui se poursuit jusqu’à ce qu’elle soit traitée avec des antibiotiques . [12] L’infection chronique des voies respiratoires est presque universelle chez les personnes atteintes de mucoviscidose, Pseudomonas aeruginosa , les champignons et les mycobactéries étant de plus en plus courants avec le temps. [13] L’inflammation des voies respiratoires supérieures entraîne un écoulement nasal fréquent et une obstruction nasale . Polypes nasauxsont fréquents, en particulier chez les enfants et les adolescents. [12] Au fur et à mesure que la maladie progresse, les gens ont tendance à avoir le souffle court et une toux chronique qui produit des expectorations . [12] Les problèmes respiratoires rendent l’exercice de plus en plus difficile, et une maladie prolongée fait que les personnes touchées souffrent d’une insuffisance pondérale pour leur âge. [12] À la fin de l’adolescence ou à l’âge adulte, les personnes commencent à développer des signes graves de maladie pulmonaire : respiration sifflante, hippocratisme digital , cyanose , crachats de sang , cardiopathie pulmonaire et affaissement pulmonaire ( atélectasie ou pneumothorax ). [12]
Dans de rares cas, la mucoviscidose peut se manifester par un trouble de la coagulation . La vitamine K est normalement absorbée par le lait maternel, le lait maternisé et, plus tard, les aliments solides. Cette absorption est altérée chez certains patients atteints de mucoviscidose. Les jeunes enfants sont particulièrement sensibles aux troubles de malabsorption de la vitamine K car seule une très petite quantité de vitamine K traverse le placenta, laissant l’enfant avec de très faibles réserves et une capacité limitée à absorber la vitamine K provenant de sources alimentaires après la naissance. Étant donné que les facteurs de coagulation II, VII, IX et X dépendent de la vitamine K, de faibles niveaux de vitamine K peuvent entraîner des problèmes de coagulation. Par conséquent, lorsqu’un enfant présente des ecchymoses inexpliquées, une évaluation de la coagulation peut être justifiée pour déterminer si une maladie sous-jacente est présente.[14]
Poumons et sinus
 Les infections respiratoires dans la mucoviscidose varient selon l’âge.
Les infections respiratoires dans la mucoviscidose varient selon l’âge.
Vert = Pseudomonas aeruginosa
Marron = Staphylococcus aureus
Bleu = Haemophilus influenzae
Rouge = Complexe Burkholderia cepacia
Les maladies pulmonaires résultent de l’obstruction des voies respiratoires due à l’accumulation de mucus, à la diminution de la clairance mucociliaire et à l’inflammation qui en résulte . [15] [16] Dans les stades ultérieurs, des changements dans l’architecture du poumon, tels que la pathologie dans les voies respiratoires principales ( bronchectasie ), exacerbent davantage les difficultés respiratoires. D’autres signes incluent une pression artérielle élevée dans les poumons ( hypertension pulmonaire ), une insuffisance cardiaque , des difficultés à fournir suffisamment d’ oxygène au corps ( hypoxie ) et une insuffisance respiratoire nécessitant une assistance avec des masques respiratoires, tels que des appareils à pression positive à deux niveaux ouventilateurs . [17] Staphylococcus aureus , Haemophilus influenzae et Pseudomonas aeruginosa sont les trois organismes les plus courants causant des infections pulmonaires chez les patients atteints de mucoviscidose. [16] : 1254 De plus, des infections opportunistes dues au complexe Burkholderia cepacia peuvent survenir, notamment par transmission de patient à patient. [18]
En plus des infections bactériennes typiques, les personnes atteintes de mucoviscidose développent plus fréquemment d’autres types de maladies pulmonaires. Parmi ceux-ci se trouve l’ aspergillose broncho-pulmonaire allergique , dans laquelle la réponse de l’organisme au champignon commun Aspergillus fumigatus provoque une aggravation des problèmes respiratoires. Une autre est l’infection par le complexe Mycobacterium avium , un groupe de bactéries liées à la tuberculose , qui peut causer des lésions pulmonaires et ne répond pas aux antibiotiques courants. [19]
Le mucus dans les sinus paranasaux est également épais et peut également provoquer un blocage des passages des sinus, entraînant une infection. Cela peut provoquer des douleurs faciales, de la fièvre, un écoulement nasal et des maux de tête . Les personnes atteintes de mucoviscidose peuvent développer une prolifération du tissu nasal (polypes nasaux ) en raison d’une inflammation due à des infections chroniques des sinus. [20] Des polypes naso-sinusiens récurrents peuvent survenir chez 10 % à 25 % des patients atteints de mucoviscidose. [16] : 1254 Ces polypes peuvent obstruer les voies nasales et augmenter les difficultés respiratoires. [21] [22]
Les complications cardiorespiratoires sont les causes de décès les plus courantes (environ 80 %) chez les patients de la plupart des centres de mucoviscidose aux États-Unis. [16] : 1254
Gastro-intestinal
En outre, la protrusion des membranes rectales internes ( prolapsus rectal ) est plus fréquente, survenant chez jusqu’à 10 % des enfants atteints de mucoviscidose, [16] et elle est causée par une augmentation du volume fécal, de la malnutrition et une augmentation de la pression intra-abdominale due à tousser. [23]
Le mucus épais observé dans les poumons a une contrepartie dans les sécrétions épaissies du pancréas , un organe chargé de fournir les sucs digestifs qui aident à décomposer les aliments. Ces sécrétions bloquent le mouvement Exocrine des enzymes digestives dans le duodénum et entraînent des lésions irréversibles du pancréas, souvent accompagnées d’une inflammation douloureuse ( pancréatite ). [24] Les canaux pancréatiques sont totalement bouchés dans les cas plus avancés, généralement observés chez les enfants plus âgés ou les adolescents. [16] Cela provoque une atrophie des glandes exocrines et une fibrose progressive. [16]
Les personnes atteintes de mucoviscidose ont également des difficultés à absorber les vitamines liposolubles A , D , E et K . [25]
En plus des problèmes de pancréas, les personnes atteintes de mucoviscidose souffrent davantage de brûlures d’ estomac , [25] d’obstruction intestinale par intussusception et de constipation . [26] Les personnes âgées atteintes de mucoviscidose peuvent développer un syndrome d’obstruction intestinale distale , qui survient lorsque les matières fécales deviennent épaisses avec du mucus ( inspissées ) et peuvent provoquer des ballonnements, des douleurs et une occlusion intestinale incomplète ou complète. [27] [25]
L’insuffisance pancréatique Exocrine survient chez la majorité (85 % à 90 %) des patients atteints de mucoviscidose. [16] : 1253 Il est principalement associé à des mutations CFTR “sévères”, où les deux allèles sont totalement non fonctionnels (ex. ΔF508 /ΔF508). [16] : 1253 Elle survient chez 10 % à 15 % des patients présentant une mutation CFTR « sévère » et une mutation « légère » où une faible activité CFTR se produit encore, ou chez lesquels deux mutations CFTR « légères » existent. [16] : 1253 Dans ces cas moins graves, une fonction Exocrine pancréatique suffisante est toujours présente pour qu’une supplémentation enzymatique ne soit pas nécessaire. [16] : 1253 Habituellement, aucune autre complication gastro-intestinale ne se produit dans les phénotypes suffisants pour le pancréas et, en général, ces personnes ont généralement une croissance et un développement excellents. [16] : 1254 Malgré cela, la pancréatite chronique idiopathique peut survenir chez un sous-ensemble d’individus atteints de mucoviscidose avec un pancréas suffisant et est associée à des douleurs abdominales récurrentes et à des complications potentiellement mortelles. [16]
Des sécrétions épaissies peuvent également causer des problèmes hépatiques chez les patients atteints de mucoviscidose. La bile sécrétée par le foie pour faciliter la digestion peut bloquer les voies biliaires , entraînant des lésions hépatiques. Une mauvaise digestion ou absorption des lipides peut entraîner une stéatorrhée . Au fil du temps, cela peut entraîner des cicatrices et une nodularité ( cirrhose ). Le foie ne parvient pas à débarrasser le sang des toxines et ne fabrique pas de protéines importantes, telles que celles responsables de la coagulation du sang . [28] [29] La maladie du foie est la troisième cause la plus fréquente de décès associée à la mucoviscidose. [16]
Environ 5 à 7 % des personnes souffrent de lésions hépatiques suffisamment graves pour provoquer des symptômes : généralement des calculs biliaires provoquant des coliques biliaires . [30]
Endocrine
Le pancréas contient les îlots de Langerhans , responsables de la fabrication de l’insuline , une hormone qui aide à réguler la glycémie . Les dommages au pancréas peuvent entraîner la perte des cellules des îlots, entraînant un type de diabète unique pour les personnes atteintes de la maladie. [31] Ce diabète lié à la mucoviscidose partage les caractéristiques des diabètes de type 1 et de type 2 et est l’une des principales complications non pulmonaires de la mucoviscidose. [32]
La vitamine D est impliquée dans la régulation du calcium et du phosphate . Une mauvaise absorption de la vitamine D provenant de l’alimentation en raison d’une malabsorption peut entraîner l’ ostéoporose , une maladie osseuse dans laquelle les os affaiblis sont plus sensibles aux fractures . [33]
Infertilité
L’infertilité touche aussi bien les hommes que les femmes. Au moins 97 % des hommes atteints de mucoviscidose sont infertiles, mais pas stériles, et peuvent avoir des enfants avec des techniques de procréation assistée. [34] La principale cause d’infertilité chez les hommes atteints de mucoviscidose est l’absence congénitale du canal déférent (qui relie normalement les testicules aux canaux éjaculateurs du pénis ), mais potentiellement aussi par d’autres mécanismes tels que l’ absence de spermatozoïdes , des spermatozoïdes de forme anormale , et peu de spermatozoïdes avec une faible motilité . [35]De nombreux hommes qui présentent une absence congénitale du canal déférent lors de l’évaluation de l’infertilité ont une forme bénigne de mucoviscidose non diagnostiquée auparavant. [36] Environ 20 % des femmes atteintes de mucoviscidose ont des difficultés de fertilité dues à un épaississement de la glaire cervicale ou à la malnutrition. Dans les cas graves, la malnutrition perturbe l’ovulation et provoque un manque de menstruation . [37]
causes
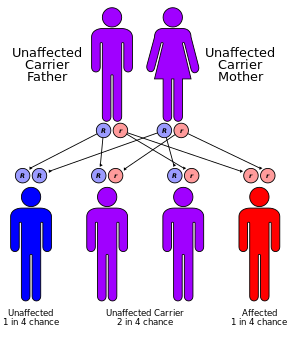 La fibrose kystique a un mode de transmission autosomique récessif
La fibrose kystique a un mode de transmission autosomique récessif
La mucoviscidose est causée par une mutation du gène régulateur de la conductance transmembranaire de la fibrose kystique ( CFTR ). La mutation la plus courante, ΔF508 , est une délétion ( Δ signifiant délétion) de trois nucléotides qui entraîne une perte de l’acide aminé phénylalanine (F) à la 508e position sur la protéine. [38] [39] Cette mutation représente les deux tiers (66 à 70 % [16] ) des cas de mucoviscidose dans le monde et 90 % des cas aux États-Unis ; cependant, plus de 1500 autres mutations peuvent produire la mucoviscidose. [40] Bien que la plupart des gens aient deux copies de travail (allèles) du CFTRgène, un seul est nécessaire pour prévenir la fibrose kystique. La FK se développe lorsqu’aucun des allèles ne peut produire une protéine CFTR fonctionnelle. Ainsi, la mucoviscidose est considérée comme une maladie autosomique récessive . [41]
Le gène CFTR , trouvé au locus q31.2 du chromosome 7 , a une longueur de 230 000 paires de bases et crée une protéine longue de 1 480 acides aminés . Plus précisément, l’emplacement se situe entre les paires de bases 117 120 016 et 117 308 718 sur le bras long du chromosome 7, région 3, bande 1, sous-bande 2, représentée par 7q31.2. Structurellement, le CFTR est un type de gène connu sous le nom de gène ABC . Le produit de ce gène (la protéine CFTR) est un canal d’ions chlorure important dans la création de sueur, de sucs digestifs et de mucus. Cette protéine possède deux domaines hydrolyseurs d’ATP , ce qui permet à la protéine d’utiliser l’énergie sous forme d’ ATP. Il contient également deux domaines comprenant chacun six hélices alpha , qui permettent à la protéine de traverser la membrane cellulaire. Un site de liaison régulateur sur la protéine permet l’activation par phosphorylation , principalement par la protéine kinase dépendante de l’AMPc . [17] Le terminal carboxyle de la protéine est ancré au cytosquelette par une interaction de domaine PDZ . [42] La majorité du CFTR dans les passages pulmonaires est produite par de rares cellules de transport d’ions qui régulent les propriétés du mucus. [43]
De plus, il est de plus en plus évident que des modificateurs génétiques autres que le CFTR modulent la fréquence et la gravité de la maladie. Un exemple est la lectine liant le mannane , qui est impliquée dans l’immunité innée en facilitant la phagocytose des micro-organismes. Les polymorphismes dans l’un ou les deux allèles de lectine liant le mannane qui entraînent des niveaux circulants inférieurs de la protéine sont associés à un risque trois fois plus élevé de maladie pulmonaire en phase terminale, ainsi qu’à un fardeau accru d’infections bactériennes chroniques. [16]
Transporteurs
Jusqu’à un individu sur 25 d’ascendance nord-européenne est considéré comme porteur génétique . La maladie n’apparaît que lorsque deux de ces porteuses ont des enfants, car chaque grossesse entre elles a 25 % de chances de produire un enfant atteint de la maladie. Bien qu’environ un nouveau-né blanc sur 3 000 soit atteint de mucoviscidose, plus de 900 mutations du gène responsable de la mucoviscidose sont connues. Les tests actuels recherchent les mutations les plus courantes. [44]
Les mutations dépistées par le test varient selon le groupe ethnique d’une personne ou selon la survenue de mucoviscidose déjà dans la famille. Plus de 10 millions d’Américains, dont un Américain blanc sur 25, sont porteurs d’une mutation du gène CF. La mucoviscidose est présente dans d’autres races, mais pas aussi fréquemment que chez les individus blancs. Environ un hispano-américain sur 46, un afro-américain sur 65 et un américain asiatique sur 90 sont porteurs d’une mutation du gène CF. [44]
Physiopathologie
 La protéine CFTR est une protéine de canal qui contrôle le flux d’ions H 2 O et Cl − entrant et sortant des cellules à l’intérieur des poumons. Lorsque la protéine CFTR fonctionne correctement, les ions entrent et sortent librement des cellules. Cependant, lorsque la protéine CFTR fonctionne mal, ces ions ne peuvent pas sortir de la cellule en raison d’un canal bloqué. Cela provoque la fibrose kystique, caractérisée par l’accumulation de mucus épais dans les poumons.
La protéine CFTR est une protéine de canal qui contrôle le flux d’ions H 2 O et Cl − entrant et sortant des cellules à l’intérieur des poumons. Lorsque la protéine CFTR fonctionne correctement, les ions entrent et sortent librement des cellules. Cependant, lorsque la protéine CFTR fonctionne mal, ces ions ne peuvent pas sortir de la cellule en raison d’un canal bloqué. Cela provoque la fibrose kystique, caractérisée par l’accumulation de mucus épais dans les poumons.
Plusieurs mutations du gène CFTR peuvent survenir et différentes mutations provoquent différents défauts de la protéine CFTR, provoquant parfois une maladie plus bénigne ou plus grave. Ces défauts protéiques sont également des cibles pour les médicaments qui peuvent parfois restaurer leur fonction. La mutation du gène ΔF508-CFTR , qui survient chez > 90 % des patients aux États-Unis, crée une protéine qui ne se replie pas normalement et n’est pas transportée de manière appropriée vers la membrane cellulaire, ce qui entraîne sa dégradation. [45]
D’autres mutations entraînent des protéines trop courtes (tronquées) car la production est arrêtée prématurément. D’autres mutations produisent des protéines qui n’utilisent pas normalement l’énergie (sous forme d’ATP), ne permettent pas au chlorure, à l’iodure et au thiocyanate de traverser la membrane de manière appropriée [46] et se dégradent à un rythme plus rapide que la normale. Les mutations peuvent également entraîner la production de moins de copies de la protéine CFTR. [17]
La protéine créée par ce gène est ancrée à la membrane externe des cellules des glandes sudoripares , des poumons, du pancréas et de toutes les autres glandes exocrines restantes dans le corps. La protéine traverse cette membrane et agit comme un canal reliant la partie interne de la cellule ( cytoplasme ) au liquide environnant . Ce canal est principalement responsable du contrôle du mouvement des anions halogénures de l’intérieur vers l’extérieur de la cellule ; cependant, dans les canaux sudoripares, il facilite le mouvement du chlorure du canal sudoripare dans le cytoplasme. Lorsque la protéine CFTR ne résorbe pas les ions dans les canaux sudoripares, le chlorure et le thiocyanate [47]libérés des glandes sudoripares sont piégés à l’intérieur des conduits et pompés vers la peau.
De plus , l’ hypothiocyanite , OSCN, ne peut pas être produit par le système de défense immunitaire. [48] [49] Parce que le chlorure est chargé négativement , cela modifie le potentiel électrique à l’intérieur et à l’extérieur de la cellule qui fait normalement passer les cations dans la cellule. Le sodium est le cation le plus courant dans l’espace extracellulaire. L’excès de chlorure dans les canaux sudoripares empêche la résorption du sodium par les canaux sodiques épithéliaux et la combinaison de sodium et de chlorure crée le sel, qui est perdu en grande quantité dans la sueur des personnes atteintes de mucoviscidose. Ce sel perdu constitue la base du test de la sueur. [17]
La plupart des dommages dans la mucoviscidose sont dus au blocage des passages étroits des organes affectés avec des sécrétions épaissies. Ces blocages entraînent un remodelage et une infection des poumons, des dommages causés par les enzymes digestives accumulées dans le pancréas, un blocage des intestins par des matières fécales épaisses, etc. Plusieurs théories ont été avancées sur la manière dont les défauts de la fonction protéique et cellulaire provoquent les effets cliniques. La théorie la plus actuelle suggère qu’un transport ionique défectueux entraîne une déshydratation de l’épithélium des voies respiratoires, épaississant le mucus. [50]Dans les cellules épithéliales des voies respiratoires, les cils existent entre la surface apicale de la cellule et le mucus dans une couche connue sous le nom de liquide de surface des voies respiratoires (ASL). Le flux d’ions de la cellule et dans cette couche est déterminé par des canaux ioniques tels que CFTR. CFTR permet non seulement aux ions chlorure d’être aspirés de la cellule et dans l’ASL, mais il régule également un autre canal appelé ENac, qui permet aux ions sodium de quitter l’ASL et d’entrer dans l’épithélium respiratoire. Le CFTR inhibe normalement ce canal, mais si le CFTR est défectueux, le sodium s’écoule librement de l’ASL et dans la cellule.
Comme l’eau suit le sodium, la profondeur de l’ASL sera épuisée et les cils seront laissés dans la couche muqueuse. [51] Comme les cils ne peuvent pas se déplacer efficacement dans un environnement épais et visqueux, la clairance mucociliaire est déficiente et une accumulation de mucus se produit, obstruant les petites voies respiratoires. [52] L’accumulation de mucus plus visqueux et riche en nutriments dans les poumons permet aux bactéries de se cacher du système immunitaire du corps, provoquant des infections respiratoires répétées. La présence des mêmes protéines CFTR dans le canal pancréatique et les glandes sudoripares de la peau provoque également des symptômes dans ces systèmes.
Infections chroniques
Les poumons des personnes atteintes de mucoviscidose sont colonisés et infectés par des bactéries dès leur plus jeune âge. Ces bactéries, qui se propagent souvent chez les personnes atteintes de mucoviscidose, se développent dans le mucus altéré, qui s’accumule dans les petites voies respiratoires des poumons. Ce mucus conduit à la formation de microenvironnements bactériens appelés biofilms difficiles à pénétrer pour les cellules immunitaires et les antibiotiques. Les sécrétions visqueuses et les infections respiratoires persistantes endommagent à plusieurs reprises le poumon en remodelant progressivement les voies respiratoires, ce qui rend l’infection encore plus difficile à éradiquer. [53] L’histoire naturelle des infections pulmonaires FK et du remodelage des voies respiratoires est mal comprise, en grande partie en raison de l’immense hétérogénéité spatiale et temporelle à la fois au sein et entre les microbiomes des patients FK. [54]
Au fil du temps, les types de bactéries et leurs caractéristiques individuelles changent chez les personnes atteintes de mucoviscidose. Au stade initial, des bactéries courantes telles que S. aureus et H. influenzae colonisent et infectent les poumons. [16] Finalement, Pseudomonas aeruginosa (et parfois Burkholderia cepacia ) domine. À 18 ans, 80 % des patients atteints de mucoviscidose classique hébergent P. aeruginosa et 3,5 % hébergent B. cepacia . [16] Une fois dans les poumons, ces bactéries s’adaptent à l’environnement et développent une résistance aux antibiotiques couramment utilisés. Pseudomonaspeut développer des caractéristiques particulières qui permettent la formation de grandes colonies, appelées Pseudomonas « mucoïdes » , rarement observées chez les personnes non atteintes de mucoviscidose. [53] Des preuves scientifiques suggèrent que la voie de l’ interleukine 17 joue un rôle clé dans la résistance et la modulation de la réponse inflammatoire lors d’une infection à P. aeruginosa dans la mucoviscidose. [55] En particulier, l’immunité médiée par l’interleukine 17 joue une activité à double tranchant lors d’une infection chronique des voies respiratoires; d’un côté, il contribue au contrôle de la charge de P. aeruginosa , tandis que de l’autre, il propage la neutrophilie pulmonaire exacerbée et le remodelage tissulaire. [55]
L’infection peut se propager en passant entre différentes personnes atteintes de mucoviscidose. [56] Par le passé, les personnes FK participaient souvent à des « camps FK » d’été et à d’autres rassemblements récréatifs. [57] [58] Les hôpitaux regroupaient les patients atteints de mucoviscidose dans des espaces communs et l’équipement de routine (tel que les nébuliseurs ) [59] n’était pas stérilisé entre les patients individuels. [60] Cela a conduit à la transmission de souches de bactéries plus dangereuses parmi des groupes de patients. En conséquence, les personnes atteintes de mucoviscidose sont désormais systématiquement isolées les unes des autres dans le cadre des soins de santé, et les prestataires de soins de santé sont encouragés à porter des blouses et des gants lors de l’examen des patients atteints de mucoviscidose afin de limiter la propagation de souches bactériennes virulentes. [61]
Les patients atteints de mucoviscidose peuvent également voir leurs voies respiratoires chroniquement colonisées par des champignons filamenteux (tels que Aspergillus fumigatus , Scedosporium apiospermum , Aspergillus terreus ) et/ou des levures (telles que Candida albicans ) ; d’autres champignons filamenteux moins couramment isolés comprennent Aspergillus flavus et Aspergillus nidulans (se produisent de manière transitoire dans les sécrétions respiratoires de la mucoviscidose) et Exophiala dermatitidis et Scedosporium prolificans (colonisateurs chroniques des voies respiratoires); certains champignons filamenteux tels que Penicillium emersonii et Acrophialophora fusisporasont rencontrées chez des patients presque exclusivement dans le cadre de la mucoviscidose. [62] La clairance mucociliaire défectueuse caractérisant la mucoviscidose est associée à des troubles immunologiques locaux. De plus, la thérapie prolongée avec des antibiotiques et l’utilisation de traitements aux corticostéroïdes peuvent également faciliter la croissance fongique. Bien que la pertinence clinique de la colonisation fongique des voies respiratoires soit encore sujette à débat, les champignons filamenteux peuvent contribuer à la réponse inflammatoire locale et donc à la détérioration progressive de la fonction pulmonaire, comme cela arrive souvent avec l’aspergillose bronchopulmonaire allergique – la maladie fongique la plus courante chez les le contexte de la mucoviscidose, impliquant une réponse immunitaire dirigée par les Th2 contre les espèces d’ Aspergillus . [62] [63]
Diagnostic
 La localisation du gène CFTR sur le chromosome 7
La localisation du gène CFTR sur le chromosome 7
Dans de nombreuses localités, tous les nouveau-nés sont soumis à un dépistage de la mucoviscidose au cours des premiers jours de leur vie, généralement par un test sanguin pour des niveaux élevés de trypsinogène immunoréactif . [64] Les nouveau-nés dont les tests sont positifs ou ceux qui sont autrement suspectés d’avoir la fibrose kystique en raison de symptômes ou d’antécédents familiaux, subissent alors un test de la sueur . Un courant électrique est utilisé pour faire pénétrer la pilocarpine dans la peau, stimulant ainsi la transpiration. La sueur est recueillie et analysée pour les niveaux de sel. Des niveaux anormalement élevés de chlorure dans la sueur suggèrent que le CFTR est dysfonctionnel ; la personne reçoit alors un diagnostic de fibrose kystique. [65] [remarque 1]Des tests génétiques sont également disponibles pour identifier les mutations CFTR généralement associées à la mucoviscidose. De nombreux laboratoires peuvent tester les 30 à 96 mutations CFTR les plus courantes, qui peuvent identifier plus de 90 % des personnes atteintes de mucoviscidose. [65]
Les personnes atteintes de mucoviscidose ont moins de thiocyanate et d’ hypothiocyanite dans leur salive [67] et leur mucus (Banfi et al.). Dans le cas de formes plus bénignes de mucoviscidose, les mesures de la différence de potentiel transépithéliale peuvent être utiles. La mucoviscidose peut également être diagnostiquée par l’identification de mutations dans le gène CFTR. [68]
Dans de nombreux cas, un parent pose le diagnostic parce que le nourrisson a un goût salé. [16] Les niveaux de trypsinogène immunoréactif peuvent être augmentés chez les individus qui ont une seule copie mutée du gène CFTR (porteurs) ou, dans de rares cas, chez les individus avec deux copies normales du gène CFTR . En raison de ces faux positifs , le dépistage de la mucoviscidose chez les nouveau-nés peut être controversé. [69] [70]
En 2010, chaque État américain avait mis en place des programmes de dépistage néonatal [71] et en 2016 [update], 21 pays européens avaient des programmes dans au moins certaines régions. [72]
Prénatal
Les femmes enceintes ou les couples planifiant une grossesse peuvent se faire tester pour les mutations du gène CFTR afin de déterminer le risque que leur enfant naisse avec la mucoviscidose. Les tests sont généralement effectués d’abord sur l’un ou les deux parents et, si le risque de mucoviscidose est élevé, les tests sur le fœtus sont effectués. L’ American College of Obstetricians and Gynecologists recommande à toutes les personnes qui envisagent de devenir enceintes de se faire tester pour voir si elles sont porteuses. [73]
Étant donné que le développement de la mucoviscidose chez le fœtus nécessite que chaque parent transmette une copie mutée du gène CFTR et que le test de mucoviscidose coûte cher, le test est souvent effectué initialement sur un parent. Si le test montre que le parent est porteur de la mutation du gène CFTR , l’autre parent est testé pour calculer le risque que ses enfants soient atteints de mucoviscidose. La mucoviscidose peut résulter de plus d’un millier de mutations différentes. [41] Depuis 2016 [update], seules les mutations les plus courantes sont généralement testées, telles que ΔF508 [41]La plupart des tests disponibles dans le commerce recherchent 32 mutations différentes ou moins. Si une famille présente une mutation rare connue, un dépistage spécifique de cette mutation peut être effectué. Parce que toutes les mutations connues ne sont pas trouvées sur les tests actuels, un dépistage négatif ne garantit pas qu’un enfant n’aura pas la mucoviscidose. [74]
Pendant la grossesse, des tests peuvent être effectués sur le placenta ( prélèvement de villosités choriales ) ou sur le liquide entourant le fœtus ( amniocentèse ). Cependant, le prélèvement de villosités choriales présente un risque de mort fœtale d’un sur 100 et d’amniocentèse d’un sur 200 ; [75] une étude récente a indiqué que cela pourrait être beaucoup plus faible, environ un sur 1 600. [76]
Sur le plan économique, pour les couples porteurs de mucoviscidose, en comparant le diagnostic génétique préimplantatoire (DPI) à la conception naturelle (NC) suivie d’un test prénatal et d’un avortement des grossesses concernées, le DPI offre des avantages économiques nets jusqu’à un âge maternel d’environ 40 ans, après quoi NC , les tests prénatals et l’avortement ont des avantages économiques plus importants. [77]
La gestion
Bien qu’aucun remède contre la mucoviscidose ne soit connu, plusieurs méthodes de traitement sont utilisées. La prise en charge de la mucoviscidose s’est considérablement améliorée au cours des 70 dernières années. Alors que les nourrissons nés avec elle il y a 70 ans auraient peu de chances de vivre au-delà de leur première année, les nourrissons d’aujourd’hui sont susceptibles de vivre jusqu’à l’âge adulte. Les progrès récents dans le traitement de la fibrose kystique ont permis aux personnes atteintes de fibrose kystique de vivre une vie plus remplie moins encombrées par leur état. Les pierres angulaires de la prise en charge sont le traitement proactif des infections des voies respiratoires et l’encouragement d’une bonne nutrition et d’un mode de vie actif. Rééducation pulmonairecar la prise en charge de la mucoviscidose se poursuit tout au long de la vie d’une personne et vise à maximiser le fonctionnement des organes, et donc la qualité de vie. Les ergothérapeutes utilisent des techniques de conservation de l’énergie (ECT) dans le processus de réadaptation des patients atteints de fibrose kystique. [78] Des exemples de techniques de conservation d’énergie sont les principes ergonomiques, la respiration des lèvres pincées et la respiration diaphragmatique. [79] Les patients atteints de mucoviscidose ont tendance à souffrir de fatigue et de dyspnée en raison d’infections pulmonaires chroniques. Par conséquent, la réduction de la quantité d’énergie dépensée pendant les activités peut aider les patients à se sentir mieux et à gagner en indépendance. [78]Au mieux, les traitements actuels retardent le déclin de la fonction des organes. En raison de la grande variation des symptômes de la maladie, le traitement a généralement lieu dans des centres multidisciplinaires spécialisés et est adapté à l’individu. Les cibles du traitement sont les poumons, le tractus gastro-intestinal (y compris les suppléments d’enzymes pancréatiques), les organes reproducteurs (y compris la technologie de procréation assistée ) et le soutien psychologique. [80]
L’aspect le plus cohérent du traitement de la mucoviscidose consiste à limiter et à traiter les lésions pulmonaires causées par un mucus épais et une infection, dans le but de maintenir la qualité de vie . Les antibiotiques intraveineux , inhalés et oraux sont utilisés pour traiter les infections chroniques et aiguës. Des dispositifs mécaniques et des médicaments par inhalation sont utilisés pour modifier et éliminer le mucus épaissi. Ces thérapies, bien qu’efficaces, peuvent prendre énormément de temps. L’oxygénothérapie à domicile est recommandée chez les personnes ayant des niveaux d’oxygène très bas. [81] De nombreuses personnes atteintes de mucoviscidose utilisent des probiotiques, qui sont censés être capables de corriger la dysbiose et l’inflammation intestinales, mais les preuves des essais cliniques concernant l’efficacité des probiotiques pour réduire les exacerbations pulmonaires chez les personnes atteintes de mucoviscidose sont incertaines. [82]
Antibiotiques
De nombreuses personnes atteintes de mucoviscidose prennent un ou plusieurs antibiotiques en tout temps, même lorsqu’elles sont en bonne santé, pour supprimer l’infection de manière prophylactique . Les antibiotiques sont absolument nécessaires chaque fois qu’une pneumonie est suspectée ou qu’un déclin notable de la fonction pulmonaire est observé, et sont généralement choisis en fonction des résultats d’une analyse des expectorations et de la réponse passée de la personne. Cette thérapie prolongée nécessite souvent une hospitalisation et l’insertion d’un intraveineux plus permanent tel qu’un cathéter central à insertion périphérique ou Port-a-Cath . Thérapie inhalée avec des antibiotiques tels que la tobramycine , la colistine et l’aztréonamest souvent administré pendant des mois pour améliorer la fonction pulmonaire en empêchant la croissance des bactéries colonisées. [83] [84] [85] L’antibiothérapie inhalée aide la fonction pulmonaire en combattant l’infection, mais présente également des inconvénients importants tels que le développement de la résistance aux antibiotiques, des acouphènes et des changements dans la voix. [86] La lévofloxacine inhalée peut être utilisée pour traiter Pseudomonas aeruginosa chez les personnes atteintes de mucoviscidose qui sont infectées. [87] La prise en charge précoce de l’infection à Pseudomonas aeruginosa est plus facile et meilleure, l’utilisation d’antibiotiques nébulisés avec ou sans antibiotiques oraux peut maintenir son éradication jusqu’à deux ans. [88]Lors du choix des antibiotiques pour traiter les patients atteints de mucoviscidose atteints d’infections pulmonaires causées par Pseudomonas aeruginosa chez les personnes atteintes de mucoviscidose, il n’est toujours pas clair si le choix des antibiotiques doit être basé sur les résultats des tests d’antibiotiques séparément (un à la fois) ou en combinaison avec chacun autre. [89]
Des antibiotiques par voie orale tels que la ciprofloxacine ou l’azithromycine sont administrés pour aider à prévenir l’infection ou à contrôler l’infection en cours. [90] Les antibiotiques aminoglycosides (par exemple la tobramycine) utilisés peuvent entraîner une perte auditive , des dommages au système d’équilibre de l’ oreille interne ou une insuffisance rénale en cas d’utilisation à long terme. [91] Pour prévenir ces effets secondaires , la quantité d’antibiotiques dans le sang est régulièrement mesurée et ajustée en conséquence. [ citation nécessaire ]
Tous ces facteurs liés à l’utilisation d’antibiotiques, à la chronicité de la maladie et à l’émergence de bactéries résistantes demandent plus d’exploration pour différentes stratégies telles que l’antibiothérapie adjuvante . [92] Actuellement, aucune preuve d’essai clinique fiable ne montre l’efficacité des antibiotiques pour les exacerbations pulmonaires chez les personnes atteintes de mucoviscidose et du complexe Burkholderia cepacia [93] ou pour l’utilisation d’antibiotiques pour traiter les mycobactéries non tuberculeuses chez les personnes atteintes de mucoviscidose. [94]
Autre médicament
Les médicaments en aérosol qui aident à déloger les sécrétions comprennent la dornase alfa et une solution saline hypertonique . [95] Dornase est une désoxyribonucléase humaine recombinante , qui décompose l’ADN dans les expectorations, diminuant ainsi sa viscosité. [96] Dornase alpha améliore la fonction pulmonaire et diminue probablement le risque d’exacerbations, mais il n’y a pas suffisamment de preuves pour savoir si elle est plus ou moins efficace que d’autres médicaments similaires. [97] La dornase alpha peut améliorer la fonction pulmonaire, mais il n’y a aucune preuve solide qu’elle soit meilleure que les autres thérapies hyperosmolaires. [97]
Le denufosol , un médicament expérimental, ouvre un canal de chlorure alternatif, aidant à liquéfier le mucus. [98] L’utilité des corticostéroïdes inhalés n’est pas claire, mais l’arrêt de la corticothérapie inhalée est sans danger. [99] Il existe de faibles preuves que le traitement aux corticostéroïdes peut causer des dommages en interférant avec la croissance. [99] La vaccination contre le pneumocoque n’a pas été étudiée en 2014 [update]. [100] En 2014 [update], il n’y a aucune preuve claire d’essais contrôlés randomisés que le vaccin contre la grippe est bénéfique pour les personnes atteintes de mucoviscidose. [101]
L’ivacaftor est un médicament administré par voie orale pour le traitement de la mucoviscidose en raison d’un certain nombre de mutations spécifiques répondant à l’amplification de la protéine CFTR induite par l’ivacaftor. [102] [103] Il améliore la fonction pulmonaire d’environ 10 % ; cependant, à partir de 2014 [update], cela coûte cher. [102] La première année qu’il était sur le marché, le prix catalogue dépassait 300 000 $ par an aux États-Unis. [102] [ nécessite une mise à jour ] En juillet 2015, la Food and Drug Administration des États-Unis a approuvé le lumacaftor/ivacaftor . [104] En 2018, la FDA a approuvé la combinaison ivacaftor/tezacaftor ; le fabricant a annoncé un prix catalogue de 292 000 $ par an. [105] Tezacaftor aide à déplacer la protéine CFTR vers la position correcte sur la surface cellulaire et est conçu pour traiter les personnes atteintes de la mutation F508del . [106]
En 2019, la combinaison médicamenteuse elexacaftor/ivacaftor/tezacaftor commercialisée sous le nom de Trikafta aux États-Unis, a été approuvée pour les patients atteints de mucoviscidose de plus de 12 ans. [107] [108] En 2021, cela a été étendu pour inclure les patients de plus de 6 ans. [109] En Europe, ce médicament a été approuvé en 2020 et commercialisé sous le nom de Kaftrio . [110] Il est utilisé chez ceux qui ont une mutation f508del, qui survient chez environ 90 % des patients atteints de mucoviscidose. [107] [111] Selon la Fondation de la fibrose kystique, “ce médicament représente la plus grande avancée thérapeutique de l’histoire de la mucoviscidose, offrant un traitement pour la cause sous-jacente de la maladie qui pourrait éventuellement apporter une thérapie modulatrice à 90 % des personnes atteintes de mucoviscidose.” [112] Dans un essai clinique, les participants qui ont reçu le médicament combiné ont connu une diminution subséquente de 63 % des exacerbations pulmonaires et une diminution de 41,8 mmol/L de la concentration de chlorure dans la sueur. [113] En atténuant un répertoire de symptômes associés à la fibrose kystique, le médicament combiné a également amélioré de manière significative les paramètres de qualité de vie chez les patients atteints de la maladie. [113] [112] Le médicament combiné est également connu pour interagir avec les inducteurs du CYP3A, comme la carbamazépine utilisée dans le traitement du trouble bipolaire, provoquant la circulation de l’elexafaftor/ivacaftor/tezacaftor dans l’organisme à des concentrations réduites. En tant que tel, l’utilisation concomitante n’est pas recommandée. [114] Le prix catalogue aux États-Unis sera de 311 000 $ par an ; [115] cependant, l’assurance peut couvrir une grande partie du coût du médicament. [116]
L’acide ursodésoxycholique , un sel biliaire , a été utilisé, mais les données sont insuffisantes pour montrer s’il est efficace. [117]
Supplémentation en nutriments
Il n’est pas certain que la supplémentation en vitamine A ou en bêta-carotène ait un effet sur les problèmes oculaires et cutanés causés par une carence en vitamine A. [118]
Il n’existe aucune preuve solide que les personnes atteintes de mucoviscidose puissent prévenir l’ostéoporose en augmentant leur apport en vitamine D. [119]
Pour les personnes souffrant de carence en vitamine E et de mucoviscidose, il existe des preuves que la supplémentation en vitamine E peut améliorer les niveaux de vitamine E, bien que l’effet de la supplémentation sur les troubles spécifiques de la carence en vitamine E ou sur la fonction pulmonaire soit encore incertain. [120]
Des preuves solides concernant les effets de la supplémentation en vitamine K chez les personnes atteintes de mucoviscidose font défaut en 2020. [121]
Diverses études ont examiné les effets de la supplémentation en acides gras oméga-3 pour les personnes atteintes de mucoviscidose, mais les preuves ne permettent pas de déterminer si elle présente des avantages ou des effets indésirables. [122]
Procédures
Plusieurs techniques mécaniques sont utilisées pour déloger les crachats et favoriser leur expectoration. Une bonne technique pour le dégagement des voies respiratoires à court terme est la physiothérapie thoracique où un inhalothérapeute percute la poitrine d’un individu à la main plusieurs fois par jour, pour relâcher les sécrétions. Cet “effet de percussion” peut également être administré par des dispositifs spécifiques qui utilisent l’ oscillation de la paroi thoracique ou un ventilateur à percussion intrapulmonaire . D’autres méthodes telles que la ventilation cuirasse biphasique , et le mode de dégagement associé disponible dans de tels dispositifs, intègrent une phase d’assistance à la toux, ainsi qu’une phase de vibration pour déloger les sécrétions. Ceux-ci sont portables et adaptés à un usage domestique. [8]
Une autre technique est la kinésithérapie par pression expiratoire positive qui consiste à fournir une contre-pression aux voies respiratoires lors de l’expiration. Cet effet est assuré par des dispositifs constitués d’un masque ou d’un embout buccal dans lesquels une résistance n’est appliquée que sur la phase d’expiration. [123] Les principes de fonctionnement de cette technique semblent être l’augmentation de la pression du gaz derrière le mucus par la ventilation collatérale ainsi qu’une augmentation temporaire de la capacité résiduelle fonctionnelle empêchant l’effondrement précoce des petites voies respiratoires lors de l’expiration. [124] [125]
À mesure que la maladie pulmonaire s’aggrave, une assistance respiratoire mécanique peut devenir nécessaire. Les personnes atteintes de mucoviscidose peuvent avoir besoin de porter des masques spéciaux la nuit pour aider à pousser l’air dans leurs poumons. Ces appareils, connus sous le nom de ventilateurs à pression positive à deux niveaux (BiPAP), aident à prévenir les faibles niveaux d’oxygène dans le sang pendant le sommeil. Des ventilateurs non invasifs peuvent être utilisés pendant la thérapie physique pour améliorer la clairance des expectorations. [126] On ne sait pas si ce type de thérapie a un impact sur les exacerbations pulmonaires ou la progression de la maladie. [126] On ne sait pas quel rôle a la thérapie de ventilation non invasive pour améliorer la capacité d’exercice chez les personnes atteintes de fibrose kystique. [126]Cependant, les auteurs ont noté que “la ventilation non invasive peut être un complément utile à d’autres techniques de dégagement des voies respiratoires, en particulier chez les personnes atteintes de mucoviscidose qui ont des difficultés à cracher des expectorations”. [127] Au cours d’une maladie grave, un tube peut être placé dans la gorge (une procédure connue sous le nom de trachéotomie ) pour permettre la respiration assistée par un ventilateur. [128] [129]
Pour les enfants, des études préliminaires montrent que la massothérapie peut améliorer la qualité de vie des personnes et de leur famille. [130]
Certaines infections pulmonaires nécessitent l’ablation chirurgicale de la partie infectée du poumon. Si cela est nécessaire plusieurs fois, la fonction pulmonaire est sévèrement réduite. [131] Les options de traitement les plus efficaces pour les personnes atteintes de mucoviscidose qui ont des pneumothorax spontanés ou récurrents ne sont pas claires. [132]
Transplantation
La transplantation pulmonaire peut devenir nécessaire pour les personnes atteintes de mucoviscidose à mesure que la fonction pulmonaire et la tolérance à l’exercice diminuent. Bien que la transplantation d’un seul poumon soit possible dans d’autres maladies, les personnes atteintes de mucoviscidose doivent faire remplacer les deux poumons car le poumon restant peut contenir des bactéries susceptibles d’infecter le poumon transplanté. Une greffe pancréatique ou hépatique peut être effectuée en même temps pour soulager une maladie du foie et/ou un diabète. [133] La transplantation pulmonaire est envisagée lorsque la fonction pulmonaire décline au point où l’assistance d’appareils mécaniques est requise ou que la survie d’une personne est menacée. [134] Selon le manuel Merck, “la transplantation pulmonaire bilatérale pour les maladies pulmonaires graves devient plus courante et plus réussie avec l’expérience et des techniques améliorées. Chez les adultes atteints de mucoviscidose, la survie médiane après la transplantation est d’environ 9 ans.” [135]
Autres aspects
 L’injection intracytoplasmique de spermatozoïdes peut être utilisée pour assurer la fertilité des hommes atteints de mucoviscidose
L’injection intracytoplasmique de spermatozoïdes peut être utilisée pour assurer la fertilité des hommes atteints de mucoviscidose
Les nouveau-nés atteints d’occlusion intestinale nécessitent généralement une intervention chirurgicale, contrairement aux adultes atteints du syndrome d’occlusion intestinale distale. Le traitement de l’insuffisance pancréatique par le remplacement des enzymes digestives manquantes permet au duodénum d’absorber correctement les nutriments et les vitamines qui seraient autrement perdus dans les selles. Cependant, la meilleure posologie et la meilleure forme de remplacement des enzymes pancréatiques ne sont pas claires, tout comme les risques et l’efficacité à long terme de ce traitement. [136]
Jusqu’à présent, aucune recherche à grande échelle portant sur l’incidence de l’ athérosclérose et des maladies coronariennes chez les adultes atteints de fibrose kystique n’a été menée. Cela est probablement dû au fait que la grande majorité des personnes atteintes de fibrose kystique ne vivent pas assez longtemps pour développer une athérosclérose ou une maladie coronarienne cliniquement significative. [137]
Le diabète est la complication non pulmonaire la plus courante de la mucoviscidose. Il mélange les caractéristiques du diabète de type 1 et de type 2 et est reconnu comme une entité distincte, le diabète lié à la mucoviscidose . [32] [138] Alors que des médicaments antidiabétiques oraux sont parfois utilisés, le traitement recommandé est l’utilisation d’ injections d’ insuline ou d’une pompe à insuline [ 139] et, contrairement au diabète de type 1 et 2, les restrictions alimentaires ne sont pas recommandées. [32] Bien que Stenotrophomonas maltophilia soit relativement fréquent chez les personnes atteintes de mucoviscidose, les preuves de l’efficacité des antibiotiques contre S. maltophilia sont incertaines.[140]
Les bisphosphonates pris par voie orale ou intraveineuse peuvent être utilisés pour améliorer la densité minérale osseuse chez les personnes atteintes de mucoviscidose. [141] Lors de la prise de bisphosphates par voie intraveineuse, les effets indésirables tels que la douleur et les symptômes pseudo-grippaux peuvent être un problème. [141] Les effets indésirables des bisphosphates pris par voie orale sur le tractus gastro-intestinal ne sont pas connus. [141]
Une mauvaise croissance peut être évitée par l’insertion d’un tube d’alimentation pour augmenter l’énergie alimentaire par des aliments supplémentaires ou par l’administration d’ hormone de croissance injectée . [142]
Les infections des sinus sont traitées par des cures prolongées d’antibiotiques. Le développement de polypes nasaux ou d’autres changements chroniques dans les voies nasales peut limiter considérablement le flux d’air par le nez et, avec le temps, réduire l’odorat de la personne. La chirurgie des sinus est souvent utilisée pour soulager l’obstruction nasale et limiter d’autres infections. Les stéroïdes nasaux tels que le propionate de fluticasone sont utilisés pour diminuer l’inflammation nasale. [143]
L’infertilité féminine peut être surmontée par la technologie de procréation assistée , en particulier les techniques de transfert d’embryons . L’infertilité masculine causée par l’absence du canal déférent peut être surmontée avec l’extraction testiculaire du sperme , en collectant les spermatozoïdes directement à partir des testicules. Si l’échantillon recueilli contient trop peu de spermatozoïdes pour avoir une fécondation spontanée, une injection intracytoplasmique de spermatozoïdes peut être effectuée. [144] La procréation par un tiers est également une possibilité pour les femmes FK. Il n’est pas clair si la prise d’ antioxydants affecte les résultats. [145]
L’exercice physique fait généralement partie des soins ambulatoires pour les personnes atteintes de mucoviscidose. [146] L’exercice aérobie semble être bénéfique pour la capacité d’exercice aérobie, la fonction pulmonaire et la qualité de vie liée à la santé ; cependant, la qualité des preuves était médiocre. [146]
En raison de l’utilisation d’antibiotiques aminoglycosides, l’ototoxicité est courante. Les symptômes peuvent inclure “acouphènes, perte auditive, hyperacousie, plénitude auditive, étourdissements et vertiges”. [147]
Gastro-intestinal
Les problèmes du système gastro-intestinal, notamment la constipation et l’obstruction du tractus gastro-intestinal, y compris le syndrome d’obstruction intestinale distale, sont des complications fréquentes chez les personnes atteintes de mucoviscidose. [27] Le traitement des problèmes gastro-intestinaux est nécessaire pour prévenir une obstruction complète, réduire les autres symptômes de la mucoviscidose et améliorer la qualité de vie. [27] Bien que les émollients fécaux, les laxatifs et les procinétiques (traitements axés sur l’IG) soient souvent suggérés, il n’y a pas de consensus clair parmi les experts quant à savoir quelle approche est la meilleure et comporte le moins de risques. [27] Des traitements mucolytiques ou systémiques visant le CFTR dysfonctionnel sont également parfois suggérés pour améliorer les symptômes. [148]
Pronostic
Le pronostic de la mucoviscidose s’est amélioré grâce à un diagnostic plus précoce grâce au dépistage, à un meilleur traitement et à un meilleur accès aux soins de santé. En 1959, l’âge médian de survie des enfants atteints de mucoviscidose aux États-Unis était de six mois. [149] En 2010, la survie est estimée à 37 ans pour les femmes et 40 ans pour les hommes. [150] Au Canada, la survie médiane est passée de 24 ans en 1982 à 47,7 en 2007. [151] Aux États-Unis, les personnes nées avec la mucoviscidose en 2016 ont une espérance de vie prévue de 47,7 ans lorsqu’elles sont soignées dans des cliniques spécialisées. [152]
Aux États-Unis, parmi les personnes atteintes de mucoviscidose âgées de plus de 18 ans en 2009, 92 % avaient obtenu leur diplôme d’études secondaires, 67 % avaient fait au moins des études universitaires, 15 % étaient handicapées, 9 % étaient au chômage, 56 % étaient célibataires. , et 39 % étaient mariés ou vivaient avec un partenaire. [153]
Qualité de vie
Les maladies chroniques peuvent être difficiles à gérer. La mucoviscidose est une maladie chronique qui affecte “les voies digestives et respiratoires entraînant une malnutrition généralisée et des infections respiratoires chroniques”. [154] Les sécrétions épaisses obstruent les voies respiratoires dans les poumons, ce qui provoque souvent une inflammation et des infections pulmonaires graves. [155] [156] S’il est compromis, il affecte la qualité de vie d’une personne atteinte de mucoviscidose et sa capacité à accomplir des tâches telles que les tâches quotidiennes.
Selon Schmitz et Goldbeck (2006), la mucoviscidose augmente considérablement le stress émotionnel de l’individu et de la famille, “et la routine de traitement quotidienne qui prend du temps peut avoir d’autres effets négatifs sur la qualité de vie”. [157] Cependant, Havermans et ses collègues (2006) ont établi que les jeunes patients ambulatoires atteints de mucoviscidose qui ont participé au questionnaire révisé sur la fibrose kystique “ont évalué certains domaines de la qualité de vie plus haut que leurs parents”. [158] Par conséquent, les patients ambulatoires atteints de mucoviscidose ont une vision plus positive d’eux-mêmes. Comme le manuel Mercknote, “avec un soutien approprié, la plupart des patients peuvent s’adapter à leur âge à la maison et à l’école. Malgré une myriade de problèmes, les réussites éducatives, professionnelles et conjugales des patients sont impressionnantes.” [135]
De plus, il existe de nombreuses façons d’améliorer la qualité de vie des patients atteints de mucoviscidose. L’exercice est promu pour augmenter la fonction pulmonaire. L’intégration d’un programme d’exercices dans la routine quotidienne du patient FK peut améliorer considérablement sa qualité de vie. [159] Aucun remède définitif pour la mucoviscidose n’est connu, mais divers médicaments sont utilisés, tels que les mucolytiques, les bronchodilatateurs, les stéroïdes et les antibiotiques, qui ont pour but de relâcher le mucus, d’élargir les voies respiratoires, de diminuer l’inflammation et de combattre les infections pulmonaires, respectivement. [160]
Épidémiologie
| Mutation | Fréquence dans le monde [161] |
|---|---|
| ΔF508 | 66 % à 70 % [16] |
| G542X | 2,4 % |
| G551D | 1,6 % |
| N1303K | 1,3 % |
| W1282X | 1,2 % |
| Tous les autres | 27,5 % |
La mucoviscidose est la maladie autosomique récessive limitant la vie la plus courante chez les personnes d’origine européenne. [162] Aux États-Unis, environ 30 000 personnes sont atteintes de mucoviscidose ; la plupart sont diagnostiqués à l’âge de six mois. Au Canada, environ 4 000 personnes sont atteintes de FK. [163] Environ 1 personne sur 25 d’origine européenne et une personne sur 30 chez les Américains blancs [164] est porteuse d’une mutation CF. Bien que la mucoviscidose soit moins fréquente dans ces groupes, environ un hispanique sur 46, un africain sur 65 et un asiatique sur 90 sont porteurs d’au moins un gène CFTR anormal . [165] [166] L’Irlande a la plus forte prévalence de mucoviscidose au monde, à un sur 1353. [167]
Bien qu’il s’agisse techniquement d’une maladie rare, la mucoviscidose est classée parmi les maladies génétiques raccourcissant la vie les plus répandues. Il est le plus courant parmi les nations du monde occidental. Une exception est la Finlande , où seulement une personne sur 80 est porteuse d’une mutation de la mucoviscidose. [168] L’ Organisation mondiale de la santé déclare: “Dans l’Union européenne, un nouveau-né sur 2 000 à 3 000 est atteint de mucoviscidose”. [169] Aux États-Unis, un enfant sur 3 500 naît avec la mucoviscidose. [170] En 1997, environ un enfant blanc sur 3 300 aux États-Unis est né avec la mucoviscidose. En revanche, seul un enfant afro-américain sur 15 000 l’a, et chez les Américains d’origine asiatique, le taux était encore plus faible à un sur 32 000. [171]
La fibrose kystique est diagnostiquée de la même manière chez les hommes et les femmes. Pour des raisons qui restent obscures, les données ont montré que les hommes ont tendance à avoir une espérance de vie plus longue que les femmes, [172] [173] bien que des études récentes suggèrent que cet écart entre les sexes pourrait ne plus exister, peut-être en raison d’améliorations dans les établissements de santé. [174] [175] Une étude récente de l’Irlande a identifié un lien entre l’oestrogène d’hormone femelle et de plus mauvais résultats dans CF. [176]
La distribution des allèles CF varie selon les populations. La fréquence des porteurs ΔF508 a été estimée à un sur 200 dans le nord de la Suède, un sur 143 en Lituanie et un sur 38 au Danemark. Aucun porteur de ΔF508 n’a été trouvé parmi 171 Finlandais et 151 Saamis . [177] ΔF508 se produit en Finlande, mais c’est un allèle minoritaire là-bas. La mucoviscidose n’est connue que dans 20 familles (arbres généalogiques) en Finlande. [178]
Évolution
On estime que la mutation ΔF508 a jusqu’à 52 000 ans. [179] De nombreuses hypothèses ont été avancées quant à la raison pour laquelle une telle mutation mortelle a persisté et s’est propagée dans la population humaine. Il a été découvert que d’autres maladies autosomiques récessives courantes telles que la drépanocytose protégeaient les porteurs d’autres maladies, un compromis évolutif connu sous le nom d’ avantage hétérozygote . La résistance aux éléments suivants a tous été proposés comme sources possibles d’avantage hétérozygote :
- Choléra : Avec la découverte que la toxine du choléra nécessite des protéines CFTR hôtes normales pour fonctionner correctement, on a émis l’hypothèse que les porteurs de gènes CFTR mutants bénéficiaient de la résistance au choléra et à d’autres causes de diarrhée. [180] [181] D’autres études n’ont pas confirmé cette hypothèse. [182] [183]
- Typhoïde : Les protéines CFTR normales sont également essentielles pour l’entrée de Salmonella Typhi dans les cellules, [184] suggérant que les porteurs de gènes CFTR mutants pourraient être résistants à la fièvre typhoïde . Aucune étude in vivo ne l’a encore confirmé. Dans les deux cas, le faible niveau de mucoviscidose en dehors de l’Europe, là où le choléra et la fièvre typhoïde sont endémiques , n’est pas immédiatement explicable.
- Diarrhée : La prévalence de la mucoviscidose en Europe pourrait être liée au développement de la domestication du bétail. Dans cette hypothèse, les porteurs d’un seul CFTR mutant avaient une certaine protection contre la diarrhée causée par l’intolérance au lactose , avant l’apparition des mutations qui ont créé la tolérance au lactose. [185]
- Tuberculose : Une autre explication possible est que les porteurs du gène pourraient avoir une certaine résistance à la tuberculose. [186] [187] Cette hypothèse est basée sur la thèse selon laquelle les porteurs de la mutation du gène CFTR ont une action insuffisante sur l’une de leurs enzymes – l’arylsulfatase – qui est nécessaire à la virulence de Mycobacterium tuberculosis . Comme M. tuberculosis utiliserait les sources de son hôte pour affecter l’individu, et en raison du manque d’enzyme, il ne pourrait pas présenter sa virulence, être porteur de la mutation CFTR pourrait fournir une résistance contre la tuberculose. [188]
Histoire
 Dorothy Hansine Andersen a décrit la mucoviscidose pour la première fois en 1938.
Dorothy Hansine Andersen a décrit la mucoviscidose pour la première fois en 1938.
La mucoviscidose est censée être apparue vers 3 000 avant JC en raison de la migration des peuples, des mutations génétiques et des nouvelles conditions d’alimentation. [189] Bien que l’ensemble du spectre clinique de la mucoviscidose n’ait été reconnu que dans les années 1930, certains aspects de la mucoviscidose ont été identifiés beaucoup plus tôt. En effet, la littérature allemande et suisse du XVIIIe siècle mettait en garde “Wehe dem Kind, das beim Kuß auf die Stirn salzig schmeckt, es ist verhext und muss bald sterben” (“Malheur à l’enfant qui a le goût salé d’un baiser sur le front, car il est maudit et doit bientôt mourir »), reconnaissant l’association entre la perte de sel dans la mucoviscidose et la maladie. [189]
Au 19e siècle, Carl von Rokitansky a décrit un cas de mort fœtale avec péritonite méconiale , une complication de l’iléus méconial associée à la mucoviscidose. L’iléus méconial a été décrit pour la première fois en 1905 par Karl Landsteiner . [189] En 1936, Guido Fanconi a décrit un lien entre la maladie cœliaque , la fibrose kystique du pancréas et la bronchectasie . [190]
En 1938, Dorothy Hansine Andersen a publié un article, “La fibrose kystique du pancréas et sa relation avec la maladie coeliaque : une étude clinique et pathologique”, dans l’ American Journal of Diseases of Children . Elle a été la première à décrire la fibrose kystique caractéristique du pancréas et à la corréler avec les maladies pulmonaires et intestinales prédominantes dans la mucoviscidose. [10] Elle a également d’abord émis l’hypothèse que la mucoviscidose était une maladie récessive et a d’abord utilisé le remplacement des enzymes pancréatiques pour traiter les enfants touchés. En 1952, Paul di Sant’Agnese a découvert des anomalies dans les électrolytes de la sueur ; un test de la sueur a été développé et amélioré au cours de la décennie suivante. [191]
Le premier lien entre la mucoviscidose et un autre marqueur (paroxonase) a été trouvé en 1985 par Hans Eiberg , indiquant qu’un seul locus existe pour la mucoviscidose. En 1988, la première mutation de la mucoviscidose, ΔF508 , a été découverte par Francis Collins , Lap-Chee Tsui et John R. Riordan sur le septième chromosome. Des recherches ultérieures ont trouvé plus de 1 000 mutations différentes qui causent la mucoviscidose.
Étant donné que les mutations du gène CFTR sont généralement petites, les techniques de génétique classique n’ont pas été en mesure d’identifier avec précision le gène muté. [192] À l’aide de marqueurs protéiques, des études de liaison génétique ont pu cartographier la mutation sur le chromosome 7. Des techniques de marche chromosomique et de saut chromosomique ont ensuite été utilisées pour identifier et séquencer le gène. [193] En 1989, Lap-Chee Tsui a dirigé une équipe de chercheurs à l’ Hospital for Sick Children de Toronto .qui a découvert le gène responsable de la mucoviscidose. La mucoviscidose représente un exemple classique de la manière dont une maladie génétique humaine a été élucidée strictement par le processus de la génétique directe .
Rechercher
Les personnes atteintes de mucoviscidose peuvent être inscrites dans un registre des maladies qui permet aux chercheurs et aux médecins de suivre les résultats de santé et d’identifier les candidats aux essais cliniques. [194]
Thérapie génique
La thérapie génique a été explorée comme remède potentiel pour la mucoviscidose. Les résultats des essais cliniques ont montré un succès limité en 2016 [update]et l’utilisation de la thérapie génique comme thérapie de routine n’est pas suggérée. [195] Une petite étude publiée en 2015 a trouvé un petit avantage. [196]
Une grande partie de la recherche sur la thérapie génique de la mucoviscidose vise à essayer de placer une copie normale du gène CFTR dans les cellules affectées. Le transfert du gène CFTR normal dans les cellules épithéliales affectées entraînerait la production de protéine CFTR fonctionnelle dans toutes les cellules cibles, sans effets indésirables ni réponse inflammatoire. Pour prévenir les manifestations pulmonaires de la mucoviscidose, seulement 5 à 10 % de la quantité normale d’expression du gène CFTR sont nécessaires. [197] Plusieurs approches ont été testées pour le transfert de gènes, telles que les liposomes et les vecteurs viraux dans des modèles animaux et des essais cliniques. Cependant, les deux méthodes se sont révélées être des options de traitement relativement inefficaces [198].principalement parce que très peu de cellules captent le vecteur et expriment le gène, de sorte que le traitement a peu d’effet. De plus, des problèmes ont été notés dans la recombinaison de l’ADNc, de sorte que le gène introduit par le traitement est rendu inutilisable. [199] Il y a eu une réparation fonctionnelle en culture de CFTR par CRISPR/Cas9 dans les organoïdes de cellules souches intestinales de patients atteints de mucoviscidose. [200]
Phagothérapie
La phagothérapie est à l’étude pour les bactéries multirésistantes chez les personnes atteintes de mucoviscidose. [201] [202]
Modulateurs de gènes
Plusieurs petites molécules visant à compenser diverses mutations du gène CFTR sont en cours de développement. Les thérapies modulatrices CFTR ont été utilisées à la place d’autres types de thérapies génétiques. Ces thérapies se concentrent sur l’expression d’une mutation génétique au lieu du gène muté lui-même. Les modulateurs sont divisés en deux classes : les potentiateurs et les correcteurs. Les potentialisateurs agissent sur les canaux ioniques CFTR qui sont intégrés dans la membrane cellulaire, et ces types de médicaments aident à ouvrir le canal pour permettre le flux transmembranaire. Les correcteurs sont destinés à aider au transport des protéines naissantes, une protéine qui est formée par les ribosomes avant de se transformer en une forme spécifique, vers la surface cellulaire pour être implémentée dans la membrane cellulaire. [203]
La plupart ciblent l’étape de transcription de l’expression génétique. Une approche a consisté à essayer de développer un médicament qui amène le ribosome à surmonter le codon d’arrêt et à produire une protéine CFTR pleine longueur. Environ 10 % des mucoviscidoses résultent d’un codon d’arrêt prématuré dans l’ADN, entraînant l’arrêt précoce de la synthèse des protéines et des protéines tronquées. Ces médicaments ciblent des mutations non-sens telles que G542X, qui se compose de l’acide aminé glycineen position 542 étant remplacé par un codon stop. Les antibiotiques aminoglycosides interfèrent avec la synthèse des protéines et la correction des erreurs. Dans certains cas, ils peuvent amener la cellule à surmonter un codon d’arrêt prématuré en insérant un acide aminé aléatoire, permettant ainsi l’expression d’une protéine de pleine longueur. Les recherches futures sur ces modulateurs se concentrent sur les cibles cellulaires qui peuvent être affectées par une modification de l’expression d’un gène. Dans le cas contraire, la thérapie génique sera utilisée comme traitement lorsque les thérapies modulatrices ne fonctionnent pas étant donné que 10 % des personnes atteintes de mucoviscidose ne sont pas concernées par ces médicaments. [204]
Elexacaftor/ivacaftor/tezacaftor a été approuvé aux États-Unis en 2019 pour la mucoviscidose. [205] Cette combinaison de médicaments précédemment développés est capable de traiter jusqu’à 90 % des personnes atteintes de mucoviscidose. [203] [205] Ce médicament restaure une certaine efficacité de la protéine CFTR afin qu’elle puisse fonctionner comme un canal ionique à la surface de la cellule. [206]
Thérapie écologique
Il a déjà été démontré que les interactions inter-espèces sont un contributeur important à la pathologie des infections pulmonaires liées à la mucoviscidose. Les exemples incluent la production d’enzymes dégradant les antibiotiques telles que les β-lactamases et la production de sous-produits métaboliques tels que les acides gras à chaîne courte (AGCC) par des espèces anaérobies, qui peuvent renforcer la pathogénicité des agents pathogènes traditionnels tels que Pseudomonas aeruginosa. [207] Pour cette raison, il a été suggéré que l’altération directe de la composition et de la fonction métabolique de la communauté microbienne de la mucoviscidose fournirait une alternative aux thérapies antibiotiques traditionnelles. [54]
Société et culture
- Sick: The Life and Death of Bob Flanagan, Supermasochist , un film documentaire de 1997
- 65 Redroses , un film documentaire de 2009
- Breathing for a Living , un mémoire de Laura Rothenberg
- Every Breath I Take, Surviving and Thriving With Cystic Fibrosis , livre de Claire Wineland
- Five Feet Apart , un film dramatique romantique de 2019 avec Cole Sprouse et Haley Lu Richardson
- Orla Tinsley: Warrior , un film documentaire de 2018 sur la militante des FC Orla Tinsley
- L’ art de la performance de Martin O’Brien
- Continent Chasers, voyageur et patient FK documentant les blogs sur les voyages et la FK, continentchasers.com
Remarques
- ^ La Cystic Fibrosis Foundation recommande un diagnostic de fibrose kystique pour toute personne suspectée de fibrose kystique (dépistage positif du nouveau-né, symptômes de fibrose kystique ou antécédents familiaux de fibrose kystique) avec un chlorure de sueur supérieur à 60 millimoles / litre. Ceux qui ont moins de 30 millimoles/litre de chlorure de sueur sont peu susceptibles de développer une fibrose kystique. Pour les personnes ayant un chlorure de sueur intermédiaire entre 30 et 59 millimoles/litre, ils recommandent des tests génétiques supplémentaires. [66]
Références
- ^ un bcdefghijklmnopqrstu O’Sullivan BP , Freedman SD ( mai 2009 ) . _ _ _ _ _ _ _ _ _ _ “Fibrose kystique”. Lancette . 373 (9678): 1891–904. doi : 10.1016/s0140-6736(09)60327-5 . PMID 19403164 . S2CID 46011502 .
- ^ Allen JL, Panitch HB, Rubenstein RC (2016). La fibrose kystique . Presse CRC. p. 92. ISBN 9781439801826. Archivé de l’original le 8 septembre 2017.
- ^ un bcd Massie J, Delatycki MB (décembre 2013). “Dépistage des porteurs de mucoviscidose”. Examens respiratoires pédiatriques . 14 (4) : 270–5. doi : 10.1016/j.prrv.2012.12.002 . PMID 23466339 .
- ^ un b Ong T, Ramsey BW (septembre 2015). “Mise à jour dans la fibrose kystique 2014”. Journal américain de médecine respiratoire et de soins intensifs . 192 (6): 669–75. doi : 10.1164/rccm.201504-0656UP . PMID 26371812 .
- ^ un bc Hodson M, Geddes D, Bush A, eds. (2012). La fibrose kystique (3e éd.). Londres : Hodder Arnold. p. 3. ISBN 978-1-4441-1369-3. Archivé de l’original le 8 septembre 2017.
- ^ Buckingham L (2012). Diagnostic moléculaire: principes fondamentaux, méthodes et applications cliniques (2e éd.). Philadelphie : FA Davis Co. p. 351.ISBN _ 978-0-8036-2975-2. Archivé de l’original le 8 septembre 2017.
- ^ Yankaskas JR, Marshall BC, Sufian B, Simon RH, Rodman D (janvier 2004). “Soins adultes de mucoviscidose : rapport de conférence de consensus” . Poitrine . 125 (1 suppl.) : 1S–39S. CiteSeerX 10.1.1.562.1904 . doi : 10.1378/chest.125.1_suppl.1S . PMID 14734689 .
- ^ un b Warnock L, Gates A (décembre 2015). “La kinésithérapie thoracique comparée à l’absence de kinésithérapie thoracique pour la mucoviscidose” . La base de données Cochrane des revues systématiques . 2015 (12) : CD001401. doi : 10.1002/14651858.CD001401.pub3 . PMC 6768986 . PMID 26688006 .
- ^ Nazareth D, Walshaw M (octobre 2013). « Passage à l’âge adulte dans la fibrose kystique – transition des soins pédiatriques aux soins pour adultes » . Médecine Clinique . 13 (5) : 482–6. doi : 10.7861/clinmedicine.13-5-482 . PMC 4953800 . PMID 24115706 .
- ^ un b Andersen DH (1938). “La fibrose kystique du pancréas et sa relation avec la maladie cœliaque : une étude clinique et pathologique”. Un m. J. Dis. Enfant. 56 (2): 344–399. doi : 10.1001/archpedi.1938.01980140114013 .
- ^ un bcd Egan , Schechter & Voynow 2020 , “Manifestations cliniques”.
- ^ un bcdef Egan , Schechter & Voynow 2020 , “ Voies respiratoires”.
- ^ Shteinberg M, Haq IJ, Polineni D, Davies JC (juin 2021). “Fibrose kystique”. Lancette . 397 (10290): 2195–2211. doi : 10.1016/S0140-6736(20)32542-3 . PMID 34090606 . S2CID 235327978 .
- ^ Reaves J, Wallace G (2010). « Ecchymoses inexpliquées : peser le pour et le contre des causes possibles » . Consultant pour Pédiatres . 9 : 201–2. Archivé de l’original le 22 février 2020 . Récupéré le 22 février 2020 .
- ^ Flume PA, Mogayzel PJ, Robinson KA, Rosenblatt RL, Quittell L, Marshall BC (août 2010). “Directives pulmonaires de mucoviscidose : complications pulmonaires : hémoptysie et pneumothorax”. Journal américain de médecine respiratoire et de soins intensifs . 182 (3): 298-306. doi : 10.1164/rccm.201002-0157OC . PMID 20675678 .
- ^ un bcdefghijklmnopqrst Mitchell RS , Kumar V , Robbins SL , et al . _ _ _ _ _ _ _ (2007). Pathologie de base de Robbins . Saunders/Elsevier. p. 1253 , 1254 . ISBN 978-1-4160-2973-1.
- ^ un bcd Rowe SM, Miller S, Sorscher EJ (mai 2005). “Fibrose kystique”. Le New England Journal of Medicine . 352 (19): 1992–2001. doi : 10.1056/NEJMra043184 . PMID 15888700 .
- ^ Saiman L, Siegel J (janvier 2004). “Contrôle de l’infection dans la mucoviscidose” . Examens de microbiologie clinique . 17 (1): 57–71. doi : 10.1128/CMR.17.1.57-71.2004 . PMC 321464 . PMID 14726455 .
- ^ Girón RM, Domingo D, Buendía B, Antón E, Ruiz-Velasco LM, Ancochea J (octobre 2005). “[Mycobactéries non tuberculeuses chez les patients atteints de mucoviscidose]”. Archivos de Bronconeumologia (en espagnol). 41 (10): 560–5. doi : 10.1016/S1579-2129(06)60283-8 . PMID 16266669 .
- ^ Franco LP, Camargos PA, Becker HM, Guimarães RE (2009). “Évaluation endoscopique nasale des enfants et des adolescents atteints de mucoviscidose” . Journal brésilien d’oto-rhino-laryngologie . 75 (6): 806–13. doi : 10.1590/S1808-86942009000600006 . PMID 20209279 .
- ^ Maldonado M, Martínez A, Alobid I, Mullol J (décembre 2004). “Le polype antrochoanal”. Rhinologie . 42 (4): 178–82. PMID 15626248 .
- ^ Ramsey B, Richardson MA (septembre 1992). “Impact de la sinusite dans la mucoviscidose”. Le Journal de l’allergie et de l’immunologie clinique . 90 (3 Pt 2): 547–52. doi : 10.1016/0091-6749(92)90183-3 . PMID 1527348 .
- ^ Kulczycki LL, Shwachman H (août 1958). « Études sur la fibrose kystique du pancréas ; apparition d’un prolapsus rectal ». Le New England Journal of Medicine . 259 (9) : 409–412. doi : 10.1056/NEJM195808282590901 . PMID 13578072 .
- ^ Cohn JA, Friedman KJ, Noone PG, Knowles MR, Silverman LM, Jowell PS (septembre 1998). “Relation entre les mutations du gène de la mucoviscidose et la pancréatite idiopathique”. Le New England Journal of Medicine . 339 (10): 653–8. doi : 10.1056/NEJM199809033391002 . PMID 9725922 .
- ^ un bc Assis DN , Freedman SD (mars 2016). “Troubles gastro-intestinaux dans la fibrose kystique”. Cliniques de médecine thoracique (révision). 37 (1): 109-118. doi : 10.1016/j.ccm.2015.11.004 . PMID 26857772 .
- ^ Malfroot A, Dab I (novembre 1991). “Nouvelles connaissances sur le reflux gastro-oesophagien dans la mucoviscidose par suivi longitudinal” . Archives des maladies de l’enfance . 66 (11): 1339-1345. doi : 10.1136/adc.66.11.1339 . PMC 1793275 . PMID 1755649 .
- ^ un bcd Carroll , Will ; Vert, Jessica ; Gilchrist, Francis J. (22 décembre 2021). “Interventions pour prévenir le syndrome d’obstruction intestinale distale (DIOS) dans la mucoviscidose” . La base de données Cochrane des revues systématiques . 12 : CD012619. doi : 10.1002/14651858.CD012619.pub3 . ISSN 1469-493X . PMC 8693853 . PMID 34936085 .
- ^ Williams SG, Westaby D, Tanner MS, Mowat AP (octobre 1992). “Problèmes hépatiques et biliaires dans la mucoviscidose”. Bulletin médical britannique . 48 (4): 877–92. doi : 10.1093/oxfordjournals.bmb.a072583 . PMID 1458306 .
- ^ Colombo C, Russo MC, Zazzeron L, Romano G (juillet 2006). “Maladie du foie dans la mucoviscidose”. Journal de gastroentérologie pédiatrique et de nutrition . 43 (Suppl 1) : S49-55. doi : 10.1097/01.mpg.0000226390.02355.52 . PMID 16819402 . S2CID 27836468 .
- ^ Egan, Schechter & Voynow 2020 , “Voies biliaires”.
- ^ Moran A, Pyzdrowski KL, Weinreb J, Kahn BB, Smith SA, Adams KS, Seaquist ER (août 1994). “La sensibilité à l’insuline dans la fibrose kystique”. Diabète . 43 (8) : 1020–6. doi : 10.2337/diabète.43.8.1020 . PMID 8039595 .
- ^ un bc de Aragão Dantas Alves C, Aguiar RA, Alves AC, Santana MA (2007) . “Le diabète sucré chez les patients atteints de mucoviscidose” . Jornal Brasileiro de Pneumologie . 33 (2): 213–221. doi : 10.1590/S1806-37132007000200017 . PMID 17724542 .
- ^ Haworth CS, Selby PL, Webb AK, Dodd ME, Musson H, McL Niven R, et al. (novembre 1999). “Faible densité minérale osseuse chez les adultes atteints de mucoviscidose” . Thorax . 54 (11): 961–7. doi : 10.1136/thx.54.11.961 . PMC 1745400 . PMID 10525552 .
- ^ McCallum TJ, Milunsky JM, Cunningham DL, Harris DH, Maher TA, Oates RD (octobre 2000). “Fertilité chez les hommes atteints de fibrose kystique: une mise à jour sur les pratiques chirurgicales actuelles et les résultats”. Poitrine . 118 (4): 1059–1062. doi : 10.1378/chest.118.4.1059 . PMID 11035677 .
- ^ Chen H, Ruan YC, Xu WM, Chen J, Chan HC (2012). “Régulation de la fertilité masculine par CFTR et implications dans l’infertilité masculine” . Mise à jour sur la reproduction humaine . 18 (6): 703–13. doi : 10.1093/humupd/dms027 . PMID 22709980 .
- ^ Augarten A, Yahav Y, Kerem BS, Halle D, Laufer J, Szeinberg A, et al. (novembre 1994). “Absence bilatérale congénitale de canal déférent en l’absence de fibrose kystique”. Lancette . 344 (8935): 1473–4. doi : 10.1016/S0140-6736(94)90292-5 . PMID 7968122 . S2CID 28860665 .
- ^ Gilljam M, Antoniou M, Shin J, Dupuis A, Corey M, Tullis DE (juillet 2000). “Grossesse dans la fibrose kystique. Résultat fœtal et maternel”. Poitrine . 118 (1): 85–91. doi : 10.1378/chest.118.1.85 . PMID 10893364 . S2CID 32289370 .
- ^ Guimbellot J, Sharma J, Rowe SM (novembre 2017). « Vers une thérapie inclusive avec les modulateurs CFTR : Progrès et défis » . Pneumologie Pédiatrique . 52 (S48) : S4–S14. doi : 10.1002/ppul.23773 . PMC 6208153 . PMID 28881097 .
- ^ Sharma J, Keeling KM, Rowe SM (août 2020). “Approches pharmacologiques pour cibler les mutations non-sens de la fibrose kystique” . Journal européen de chimie médicinale . 200 : 112436. doi : 10.1016/j.ejmech.2020.112436 . PMC 7384597 . PMID 32512483 .
- ^ Bobadilla JL, Macek M, Fine JP, Farrell PM (juin 2002). “Mucoviscidose : une analyse mondiale des mutations CFTR – corrélation avec les données d’incidence et application au dépistage” . Mutations Humaines . 19 (6): 575–606. doi : 10.1002/humu.10041 . PMID 12007216 . S2CID 35428054 .
- ^ un bc Elborn JS (novembre 2016). “Fibrose kystique”. Lancette . 388 (10059): 2519–2531. doi : 10.1016/S0140-6736(16)00576-6 . PMID 27140670 . S2CID 20948144 .
- ^ Short DB, Trotter KW, Reczek D, Kreda SM, Bretscher A, Boucher RC, et al. (juillet 1998). “Une protéine PDZ apicale ancre le régulateur de conductance transmembranaire de la fibrose kystique au cytosquelette” . Le Journal de chimie biologique . 273 (31): 19797–801. doi : 10.1074/jbc.273.31.19797 . PMID 9677412 .
- ^ Travaglini KJ, Krasnow MA (août 2018). “Profil d’une cellule respiratoire inconnue” . Nature . 560 (7718): 313–314. Bibcode : 2018Natur.560..313T . doi : 10.1038/d41586-018-05813-7 . PMID 30097657 .
- ^ un b Edwards QT, Seibert D, Macri C, Covington C, Tilghman J (novembre 2004). “Évaluer l’ethnicité dans le conseil préconceptionnel: génétique – ce que les infirmières praticiennes doivent savoir”. Journal de l’Académie américaine des infirmières praticiennes . 16 (11): 472–80. doi : 10.1111/j.1745-7599.2004.tb00426.x . PMID 15617360 . S2CID 7644129 .
- ^ Wang, Xiaodong Robert; Li, Chenglong (6 mai 2014). “Décodage du mauvais repliement F508del dans la fibrose kystique” . Biomolécules . 4 (2): 498–509. doi : 10.3390/biom4020498 . ISSN 2218-273X . PMC 4101494 . PMID 24970227 .
- ^ Childers M, Eckel G, Himmel A, Caldwell J (2007). “Un nouveau modèle de pathologie de la mucoviscidose : manque de transport du glutathion et de ses conjugués thiocyanate”. Hypothèses médicales . 68 (1): 101–12. doi : 10.1016/j.mehy.2006.06.020 . PMID 16934416 .
- ^ Xu Y, Szép S, Lu Z (décembre 2009). “Le rôle antioxydant du thiocyanate dans la pathogenèse de la mucoviscidose et d’autres maladies liées à l’inflammation” . Actes de l’Académie nationale des sciences des États-Unis d’Amérique . 106 (48): 20515–9. Bibcode : 2009PNAS..10620515X . doi : 10.1073/pnas.0911412106 . PMC 2777967 . PMID 19918082 .
- ^ Moskwa P, Lorentzen D, Excoffon KJ, Zabner J, McCray PB, Nauseef WM, et al. (janvier 2007). “Un nouveau système de défense de l’hôte des voies respiratoires est défectueux dans la mucoviscidose” . Journal américain de médecine respiratoire et de soins intensifs . 175 (2): 174–183. doi : 10.1164/rccm.200607-1029OC . PMC 2720149 . PMID 17082494 .
- ^ Conner GE, Wijkstrom-Frei C, Randell SH, Fernandez VE, Salathe M (janvier 2007). “Le système lactoperoxydase relie le transport des anions à la défense de l’hôte dans la mucoviscidose” . Lettres FEBS . 581 (2): 271–8. doi : 10.1016/j.febslet.2006.12.025 . PMC 1851694 . PMID 17204267 .
- ^ Haq IJ, Gray MA, Garnett JP, Ward C, Brodlie M (mars 2016). « Homéostasie liquidienne de surface des voies respiratoires dans la mucoviscidose : physiopathologie et cibles thérapeutiques » . Thorax . 71 (3): 284–287. doi : 10.1136/thoraxjnl-2015-207588 . PMID 26719229 .
- ^ Verkman AS, Song Y, Thiagarajah JR (janvier 2003). “Rôle du liquide de surface des voies respiratoires et des glandes sous-muqueuses dans la maladie pulmonaire de la fibrose kystique”. Journal américain de physiologie. Physiologie cellulaire . 284 (1) : C2-15. doi : 10.1152/ajpcell.00417.2002 . PMID 12475759 . S2CID 11790119 .
- ^ Marieb EN, Hoehn K, Hutchinson M (2014). “22 : Le système respiratoire”. Anatomie et physiologie humaines . Éducation Pearson. p. 906.ISBN _ 978-0805361179.
- ^ un b Saiman L (2004). « Microbiologie des maladies pulmonaires précoces de la mucoviscidose ». Examens respiratoires pédiatriques . 5 (supplément A) : S367-9. doi : 10.1016/S1526-0542(04)90065-6 . PMID 14980298 .
- ^ un b Khanolkar RA, Clark ST, Wang PW, et autres. (2020). “Succession écologique des communautés polymicrobiennes dans les voies respiratoires de la fibrose kystique” . mSystèmes . 5 (6) : e00809-20. doi : 10.1128/mSystems.00809-20 . PMC 7716390 . PMID 33262240 .
- ^ un b Lorè NI, Cigana C, Riva C, De Fino I, Nonis A, Spagnuolo L, et al. (mai 2016). “L’IL-17A altère la tolérance de l’hôte lors d’une infection chronique des voies respiratoires par Pseudomonas aeruginosa” . Rapports scientifiques . 6 : 25937. Bibcode : 2016NatSR…625937L . doi : 10.1038/srep25937 . PMC 4870500 . PMID 27189736 .
- ^ Tümmler B, Koopmann U, Grothues D, Weissbrodt H, Steinkamp G, von der Hardt H (juin 1991). “Acquisition nosocomiale de Pseudomonas aeruginosa par des patients atteints de mucoviscidose” . Journal de microbiologie clinique . 29 (6) : 1265–7. Bibcode : 1991JPoSA..29.1265A . doi : 10.1002/pola.1991.080290905 . PMC 271975 . PMID 1907611 .
- ^ Centres pour la prévention du contrôle des maladies (CDC) (juin 1993). “Pseudomonas cepacia dans les camps d’été pour les personnes atteintes de fibrose kystique”. MMWR. Rapport hebdomadaire sur la morbidité et la mortalité . 42 (23): 456–9. PMID 7684813 .
- ^ Pegues DA, Carson LA, Tablan OC, FitzSimmons SC, Roman SB, Miller JM, Jarvis WR (mai 1994). “Acquisition de Pseudomonas cepacia dans les camps d’été pour les patients atteints de fibrose kystique. Groupe d’étude des camps d’été” . Le Journal de pédiatrie . 124 (5 Pt 1): 694–702. doi : 10.1016/S0022-3476(05)81357-5 . PMID 7513755 .
- ^ Pankhurst CL, Philpott-Howard J (avril 1996). “Les facteurs de risque environnementaux associés à l’équipement médical et dentaire dans la transmission de Burkholderia (Pseudomonas) cepacia chez les patients atteints de mucoviscidose”. Le Journal des infections hospitalières . 32 (4) : 249–255. doi : 10.1016/S0195-6701(96)90035-3 . PMID 8744509 .
- ^ Jones AM, Govan JR, Doherty CJ, Dodd ME, Isalska BJ, Stanbridge TN, Webb AK (juin 2003). “Identification de la diffusion aérienne de souches multirésistantes épidémiques de Pseudomonas aeruginosa dans un centre de mucoviscidose lors d’une épidémie d’infection croisée” . Thorax . 58 (6) : 525–7. doi : 10.1136/thorax.58.6.525 . PMC 1746694 . PMID 12775867 .
- ^ Høiby N (juin 1995). “Isolement et traitement des patients atteints de fibrose kystique atteints d’infections pulmonaires causées par Pseudomonas (Burkholderia) cepacia et Pseudomonas aeruginosa multirésistant”. Le Journal néerlandais de médecine . 46 (6) : 280–7. doi : 10.1016/0300-2977(95)00020-N . PMID 7643943 .
- ^ un b Pihet M, Carrère J, Cimon B, Chabasse D, Delhaes L, Symoens F, Bouchara JP (juin 2009). “Occurrence et pertinence des champignons filamenteux dans les sécrétions respiratoires des patients atteints de fibrose kystique – une revue” . Mycologie médicale . 47 (4): 387–397. doi : 10.1080/13693780802609604 . PMID 19107638 .
- ^ Rapaka RR, Kolls JK (2009). “Pathogenèse de l’aspergillose broncho-pulmonaire allergique dans la mucoviscidose : compréhension actuelle et orientations futures” . Mycologie médicale . 47 (Suppl 1) : S331-7. doi : 10.1080/13693780802266777 . PMID 18668399 .
- ^ “Dépistage néonatal pour CF” . Fondation de la fibrose kystique . Récupéré le 25 janvier 2022 .
- ^ un b Egan, Schechter & Voynow 2020 , “Diagnostic et évaluation”.
- ^ Farrell PM, White TB, Ren CL, Hempstead SE, Accurso F, Derichs N, Howenstine M, McColley SA, Rock M, Rosenfeld M, Sermet-Gaudelus I, Southern KW, Marshall BC, Sosnay PR (février 2017). “Diagnostic de mucoviscidose : Directives de consensus de la base de mucoviscidose”. J Pédiatre . 181S : S4–S15.e1. doi : 10.1016/j.jpeds.2016.09.064 . PMID 28129811 . S2CID 206410545 .
- ^ Minarowski Ł, Sands D, Minarowska A, Karwowska A, Sulewska A, Gacko M, Chyczewska E (2008). “Concentration de thiocyanate dans la salive des patients atteints de mucoviscidose” . Folia Histochemica et Cytobiologica . 46 (2): 245–6. doi : 10.2478/v10042-008-0037-0 . PMID 18519245 .
- ^ Stern RC (février 1997). “Le diagnostic de la mucoviscidose” . Le New England Journal of Medicine . 336 (7): 487–91. doi : 10.1056/NEJM199702133360707 . PMID 9017943 .
- ^ Ross LF (septembre 2008). « Dépistage néonatal de la mucoviscidose : une leçon sur les disparités de santé publique » . Le Journal de pédiatrie . 153 (3): 308–313. doi : 10.1016/j.jpeds.2008.04.061 . PMC 2569148 . PMID 18718257 .
- ^ Assael BM, Castellani C, Ocampo MB, Iansa P, Callegaro A, Valsecchi MG (septembre 2002). “Analyse de l’épidémiologie et de la survie de la mucoviscidose dans une zone de dépistage néonatal intense sur 30 ans” . Journal américain d’épidémiologie . 156 (5): 397–401. doi : 10.1093/aje/kwf064 . PMID 12196308 .
- ^ Hoch H, Sontag MK, Scarbro S, Juarez-Colunga E, McLean C, Kempe A, Sagel SD (novembre 2018). “Résultats cliniques chez les nourrissons américains atteints de fibrose kystique de 2001 à 2012”. Pneumologie Pédiatrique . 53 (11): 1492–1497. doi : 10.1002/ppul.24165 . PMID 30259702 . S2CID 52845580 .
- ^ Barben, Jürg, et al. (2017). “L’expansion et la performance des programmes nationaux de dépistage néonatal de la mucoviscidose en Europe”. Journal de la fibrose kystique . 16 (2): 207–213. doi : 10.1016/j.jcf.2016.12.012 . PMID 28043799 .
- ^ “Dépistage des porteurs à l’ère de la médecine génomique” . Collège américain des obstétriciens et gynécologues . 2017. Archivé de l’original le 25 février 2017 . Récupéré le 22 février 2020 .
- ^ Elias S, Annas GJ, Simpson JL (avril 1991). “Dépistage des porteurs de la fibrose kystique : implications pour la pratique obstétricale et gynécologique”. Journal américain d’obstétrique et de gynécologie . 164 (4): 1077–1083. doi : 10.1016/0002-9378(91)90589-j . PMID 2014829 .
- ^ Tabor A, Philip J, Madsen M, Bang J, Obel EB, Nørgaard-Pedersen B (juin 1986). “Essai contrôlé randomisé d’amniocentèse génétique chez 4606 femmes à faible risque”. Lancette . 1 (8493): 1287–93. doi : 10.1016/S0140-6736(86)91218-3 . PMID 2423826 . S2CID 31237495 .
- ^ Eddleman KA, Malone FD, Sullivan L, Dukes K, Berkowitz RL, Kharbutli Y, et al. (novembre 2006). “Taux de perte de grossesse après amniocentèse au milieu du trimestre”. Obstétrique et Gynécologie . 108 (5): 1067-1072. doi : 10.1097/01.AOG.0000240135.13594.07 . PMID 17077226 . S2CID 19081825 .
- ^ Davis LB, Champion SJ, Fair SO, Baker VL, Garber AM (avril 2010). “Une analyse coûts-avantages du diagnostic génétique préimplantatoire pour les couples porteurs de la mucoviscidose”. Fertilité et stérilité . 93 (6): 1793–804. doi : 10.1016/j.fertnstert.2008.12.053 . PMID 19439290 .
- ^ un b “Résumés du 25e Congrès italien de la fibrose kystique et du 15e Congrès national de la Société italienne de la fibrose kystique : Assago, Milan. 10 – 12 octobre 2019” . Journal italien de pédiatrie . 46 (Suppl 1) : 32 avril 2020. doi : 10.1186/s13052-020-0790-z . PMC 7110616 . PMID 32234058 .
- ^ Wingårdh AS, Göransson C, Larsson S, Slinde F, Vanfleteren LE (2020). “Efficacité des techniques de conservation de l’énergie chez les patients atteints de MPOC” . Respiration; Revue internationale des maladies thoraciques . 99 (5): 409–416. doi : 10.1159/000506816 . PMC 7265758 . PMID 32272478 .
- ^ Davies JC, Alton EW, Bush A (décembre 2007). “Mucoviscidose” . BMJ . 335 (7632): 1255–9. doi : 10.1136/bmj.39391.713229.AD . PMC 2137053 . PMID 18079549 .
- ^ Hayes D, Wilson KC, Krivchenia K, Hawkins SM, Balfour-Lynn IM, Gozal D, et al. (février 2019). “L’oxygénothérapie à domicile pour les enfants. Un guide officiel de pratique clinique de l’American Thoracic Society” . Journal américain de médecine respiratoire et de soins intensifs . 199 (3) : e5–e23. doi : 10.1164/rccm.201812-2276ST . PMC 6802853 . PMID 30707039 .
- ^ Coffey MJ, Garg M, Homaira N, Jaffe A, Ooi CY (janvier 2020). “Probiotiques pour les personnes atteintes de mucoviscidose” . La base de données Cochrane des revues systématiques . 1 : CD012949. doi : 10.1002/14651858.CD012949.pub2 . PMC 6984633 . PMID 31962375 .
- ^ Pai VB, Nahata MC (octobre 2001). “Efficacité et sécurité de la tobramycine en aérosol dans la fibrose kystique”. Pneumologie Pédiatrique . 32 (4) : 314–327. doi : 10.1002/ppul.1125 . PMID 11568993 . S2CID 30108514 .
- ^ Westerman EM, Le Brun PP, Touw DJ, Frijlink HW, Heijerman HG (mars 2004). “Effet du sulfate de colistine nébulisé et du sulfométhate de colistine sur la fonction pulmonaire chez les patients atteints de mucoviscidose : une étude pilote” . Journal de la fibrose kystique . 3 (1) : 23-8. doi : 10.1016/j.jcf.2003.12.005 . PMID 15463883 .
- ^ McCoy KS, Quittner AL, Oermann CM, Gibson RL, Retsch-Bogart GZ, Montgomery AB (novembre 2008). “Lysine d’aztréonam inhalée pour les voies respiratoires chroniques Pseudomonas aeruginosa dans la fibrose kystique” . Journal américain de médecine respiratoire et de soins intensifs . 178 (9) : 921–8. doi : 10.1164/rccm.200712-1804OC . PMC 2577727 . PMID 18658109 .
- ^ Ryan G, Singh M, Dwan K (mars 2011). “Antibiotiques inhalés pour la thérapie à long terme dans la mucoviscidose”. La base de données Cochrane des revues systématiques (3) : CD001021. doi : 10.1002/14651858.CD001021.pub2 . PMID 21412868 .
- ^ “Quinsair (lévofloxacine)” . Agence européenne des médicaments . Archivé de l’original le 26 décembre 2016 . Récupéré le 26 décembre 2016 .
- ^ Langton Hewer SC, Smyth AR (avril 2017). “Stratégies antibiotiques pour éradiquer Pseudomonas aeruginosa chez les personnes atteintes de mucoviscidose” (PDF) . La base de données Cochrane des revues systématiques . 4 (4) : CD004197. doi : 10.1002/14651858.CD004197.pub5 . PMC 6478104 . PMID 28440853 . Archivé de l’original (PDF) le 24 février 2019 . Récupéré le 22 janvier 2019 .
- ^ Smith S, Ratjen F, Remmington T, Waters V (mai 2020). “Tests de sensibilité aux antimicrobiens combinés pour les exacerbations aiguës dans l’infection chronique de Pseudomonas aeruginosa dans la fibrose kystique” . La base de données Cochrane des revues systématiques . 5 (4) : CD006961. doi : 10.1002/14651858.CD006961.pub5 . PMC 7387858 . PMID 32412092 .
- ^ Hansen CR, Pressler T, Koch C, Høiby N (mars 2005). “Traitement à long terme par l’azitromycine des patients atteints de fibrose kystique atteints d’une infection chronique à Pseudomonas aeruginosa ; une étude de cohorte observationnelle” . Journal de la fibrose kystique . 4 (1): 35–40. doi : 10.1016/j.jcf.2004.09.001 . PMID 15752679 .
- ^ Tan KH, Mulheran M, Knox AJ, Smyth AR (mars 2003). « Prescription et surveillance des aminoglycosides dans la fibrose kystique ». Journal américain de médecine respiratoire et de soins intensifs . 167 (6): 819–23. doi : 10.1164/rccm.200109-012CC . PMID 12623858 .
- ^ Hurley MN, Smith S, Forrester DL, Smyth AR (juillet 2020). “Traitement adjuvant antibiotique pour l’infection pulmonaire dans la fibrose kystique” . La base de données Cochrane des revues systématiques . 7 (9) : CD008037. doi : 10.1002/14651858.CD008037.pub4 . PMC 8407502 . PMID 32671834 .
- ^ Lord R, Jones AM, Horsley A (avril 2020). “Traitement antibiotique du complexe Burkholderia cepacia chez les personnes atteintes de mucoviscidose souffrant d’une exacerbation pulmonaire” . La base de données Cochrane des revues systématiques . 2020 (4) : CD009529. doi : 10.1002/14651858.CD009529.pub4 . PMC 7117566 . PMID 32239690 .
- ^ Waters V, Ratjen F (juin 2020). “Traitement antibiotique de l’infection pulmonaire à mycobactéries non tuberculeuses chez les personnes atteintes de mucoviscidose” . La base de données Cochrane des revues systématiques . 6 : CD010004. doi : 10.1002/14651858.CD010004.pub5 . PMC 7389742 . PMID 32521055 .
- ^ Kuver R, Lee SP (avril 2006). “Saline hypertonique pour la mucoviscidose”. Le New England Journal of Medicine . 354 (17): 1848–51, réponse de l’auteur 1848–51. doi : 10.1056/NEJMc060351 . PMID 16642591 .
- ^ Lieberman J (juillet 1968). “Effet d’aérosol Dornase sur la viscosité des expectorations dans les cas de fibrose kystique”. JAMA . 205 (5) : 312–3. doi : 10.1001/jama.205.5.312 . PMID 5694947 .
- ^ un b Yang C, Montgomery M, et autres. (Groupe Cochrane sur la mucoviscidose et les autres maladies génétiques) (mars 2021). “Dornase alfa pour la mucoviscidose” . La base de données Cochrane des revues systématiques . 2021 (3) : CD001127. doi : 10.1002/14651858.CD001127.pub5 . PMC 8094421 . PMID 33735508 .
- ^ Kellerman D, Rossi Mospan A, Engels J, Schaberg A, Gorden J, Smiley L (août 2008). “Denufosol: un examen des études avec des agonistes P2Y (2) inhalés qui ont conduit à la phase 3”. Pharmacologie pulmonaire et thérapeutique . 21 (4) : 600–7. doi : 10.1016/j.pupt.2007.12.003 . PMID 18276176 .
- ^ un b Balfour-Lynn IM, Welch K, Smith S (juillet 2019). “Corticostéroïdes inhalés pour la mucoviscidose” . La base de données Cochrane des revues systématiques . 7 (4) : CD001915. doi : 10.1002/14651858.CD001915.pub6 . PMC 6609325 . PMID 31271656 .
- ^ Burgess L, Southern KW (août 2014). Burgess L (éd.). “Vaccins antipneumococciques pour la mucoviscidose”. La base de données Cochrane des revues systématiques . 8 (8) : CD008865. doi : 10.1002/14651858.CD008865.pub3 . PMID 25093421 .
- ^ Dharmaraj P, Smyth RL (mars 2014). “Vaccins pour prévenir la grippe chez les personnes atteintes de mucoviscidose” . La base de données Cochrane des revues systématiques . 2021 (3) : CD001753. doi : 10.1002/14651858.CD001753.pub3 . PMC 7066935 . PMID 24604671 .
- ^ un bc Whiting P, Al M, Burgers L, Westwood M, Ryder S, Hoogendoorn M, et al. (Mars 2014). “Ivacaftor pour le traitement des patients atteints de mucoviscidose et de la mutation G551D : une revue systématique et une analyse coût-efficacité” . Évaluation des technologies de la santé . 18 (18): 1–106. doi : 10.3310/hta18180 . PMC 4780965 . PMID 24656117 .
- ^ Wainwright CE (octobre 2014). “Ivacaftor pour les patients atteints de mucoviscidose”. Examen d’experts en médecine respiratoire . 8 (5) : 533–8. doi : 10.1586/17476348.2014.951333 . PMID 25148205 . S2CID 39537446 .
- ^ Bureau du commissaire. “Annonces de presse – La FDA approuve un nouveau traitement pour la fibrose kystique” . www.fda.gov . Archivé de l’original le 18 janvier 2017 . Récupéré le 16 janvier 2017 .
- ^ “La FDA approuve un autre médicament Vertex pour le traitement de la fibrose kystique – Le Boston Globe” . Le BostonGlobe .
- ^ “Tezacaftor (VX-661) pour la fibrose kystique – Nouvelles de la fibrose kystique aujourd’hui” . Archivé de l’original le 29 septembre 2018 . Récupéré le 23 décembre 2018 .
- ^ un b “l’Histoire d’Approbation de FDA de Trikafta (elexacaftor, ivacaftor et tezacaftor)” . Drugs.com .
- ^ Commissaire, Bureau du (24 mars 2020). “La FDA approuve une nouvelle thérapie révolutionnaire pour la mucoviscidose” . FDA . Récupéré le 28 avril 2022 .
- ^ “La FDA accepte l’application Vertex pour l’expansion de Trikafta pour inclure les enfants âgés de 6 à 11 ans | Fondation de la fibrose kystique” . www.cff.org . Récupéré le 28 avril 2022 .
- ^ “NHS Angleterre” Accord historique du NHS pour ouvrir l’accès à un médicament contre la fibrose kystique qui change la vie” . www.angleterre.nhs.uk . Récupéré le 8 août 2021 .
- ^ Bureau du commissaire (24 mars 2020). “La FDA approuve une nouvelle thérapie révolutionnaire pour la mucoviscidose” . FDA . Récupéré le 12 août 2020 .
- ^ un b “Déclaration de la Fondation de la fibrose kystique sur l’approbation par la FDA de Trikafta TM, la première thérapie à triple combinaison pour la mutation CF la plus courante” . www.cff.org . Bethesda, Md. : Fondation de la fibrose kystique . Récupéré le 12 août 2020 .
- ^ a b Middleton PG, Mall MA, Dřevínek P, Lands LC, McKone EF, Polineni D, et al. (novembre 2019). “Elexacaftor-Tezacaftor-Ivacaftor pour la fibrose kystique avec un seul allèle Phe508del” . Le New England Journal of Medicine . 381 (19): 1809-1819. doi : 10.1056/NEJMoa1908639 . PMC 7282384 . PMID 31697873 .
- ^ Ridley K, Condren M (1er avril 2020). “Elexacaftor-Tezacaftor-Ivacaftor : la première thérapie de modulation du régulateur de conductance transmembranaire de la fibrose kystique à triple combinaison” . Le Journal de pharmacologie pédiatrique et thérapeutique . 25 (3): 192–197. doi : 10.5863/1551-6776-25.3.192 . PMC 7134581 . PMID 32265602 .
- ^ “Vertex évalue le traitement combiné de la fibrose kystique à 311 000 $ par an” . Reuters . 21 octobre 2019 . Récupéré le 23 octobre 2019 .
- ^ Réseau de bonnes nouvelles (3 novembre 2019). “La FDA approuve le premier nouveau traitement de la fibrose kystique depuis des décennies” . Réseau Bonnes Nouvelles . Récupéré le 12 août 2020 .
- ^ Cheng K, Ashby D, Smyth RL (septembre 2017). “Acide ursodésoxycholique pour les maladies hépatiques liées à la fibrose kystique” . La base de données Cochrane des revues systématiques . 9 (4) : CD000222. doi : 10.1002/14651858.CD000222.pub4 . PMC 6483662 . PMID 28891588 .
- ^ de Vries JJ, Chang AB, Bonifant CM, Shevill E, Marchant JM (août 2018). “Supplémentation en vitamine A et bêta (β)-carotène pour la mucoviscidose” . La base de données Cochrane des revues systématiques . 8 : CD006751. doi : 10.1002/14651858.CD006751.pub5 . PMC 6513379 . PMID 30091146 .
- ^ Ferguson JH, Chang AB (mai 2014). “Supplémentation en vitamine D pour la mucoviscidose”. La base de données Cochrane des revues systématiques (5) : CD007298. doi : 10.1002/14651858.CD007298.pub4 . PMID 24823922 .
- ^ Okebukola PO, Kansra S, Barrett J (septembre 2020). “Supplémentation en vitamine E chez les personnes atteintes de mucoviscidose” . La base de données Cochrane des revues systématiques . 2020 (9) : CD009422. doi : 10.1002/14651858.CD009422.pub4 . PMC 8406985 . PMID 32892350 .
- ^ Jagannath VA, Thaker V, Chang AB, Price AI (juin 2020). “Supplémentation en vitamine K pour la mucoviscidose” . La base de données Cochrane des revues systématiques . John Wiley & fils, Ltd. 6 (6) : CD008482. doi : 10.1002/14651858.CD008482.pub6 . PMC 7272115 . PMID 32497260 .
- ^ Watson H, Stackhouse C (avril 2020). “Supplémentation en acides gras oméga-3 pour la fibrose kystique” . La base de données Cochrane des revues systématiques . 4 : CD002201. doi : 10.1002/14651858.CD002201.pub6 . PMC 7147930 . PMID 32275788 .
- ^ McIlwaine M, Bouton B, Nevitt SJ (novembre 2019). “Physiothérapie par pression expiratoire positive pour le dégagement des voies respiratoires chez les personnes atteintes de mucoviscidose” . La base de données Cochrane des revues systématiques . 2019 (11). doi : 10.1002/14651858.CD003147.pub5 . PMC 6953327 . PMID 31774149 .
- ^ Andersen JB, Qvist J, Kann T (octobre 1979). “Recruter le poumon effondré par les canaux collatéraux avec une pression positive en fin d’expiration”. Journal scandinave des maladies respiratoires . 60 (5) : 260–6. PMID 392747 .
- ^ Groth S, Stafanger G, Dirksen H, Andersen JB, Falk M, Kelstrup M (juillet 1985). “La physiothérapie par pression expiratoire positive (PEP-masque) améliore la ventilation et réduit le volume de gaz piégé dans la mucoviscidose”. Bulletin Européen de Physiopathologie Respiratoire . 21 (4): 339–343. PMID 3899222 .
- ^ un bc Moran F, Bradley JM, Piper AJ (février 2017). “Ventilation non invasive pour la mucoviscidose” . La base de données Cochrane des revues systématiques . 2 : CD002769. doi : 10.1002/14651858.CD002769.pub5 . PMC 6464053 . PMID 28218802 .
- ^ Moran F, Bradley JM, Piper AJ (février 2017). “Ventilation non invasive pour la mucoviscidose” . La base de données Cochrane des revues systématiques . 2 (2) : CD002769. doi : 10.1002/14651858.CD002769.pub5 . PMC 6464053 . PMID 28218802 .
- ^ “Trachéotomie Pourquoi est-il utilisé” . NHS. 3 octobre 2018 . Récupéré le 10 mai 2020 .
- ^ Molnar H. “Types de tubes de trachéotomie” .
- ^ Huth MM, Zink KA, Van Horn NR (2005). “Les effets de la massothérapie dans l’amélioration des résultats pour les jeunes atteints de fibrose kystique : un examen des preuves”. Soins infirmiers pédiatriques . 31 (4): 328-32. PMID 16229132 .
- ^ Leinwand MJ (28 décembre 2019). Windle ML, Odim J (éd.). “Traitement chirurgical des infections du poumon, de la plèvre et du médiastin” . Medscape . Archivé de l’original le 5 octobre 2016.
- ^ Amin R, Noone PG, Ratjen F (décembre 2012). “Pleurodèse chimique versus intervention chirurgicale pour les pneumothorax persistants et récurrents dans la mucoviscidose” . La base de données Cochrane des revues systématiques . 12 : CD007481. doi : 10.1002/14651858.CD007481.pub3 . PMC 7208277 . PMID 23235645 .
- ^ Fridell JA, Vianna R, Kwo PY, Howenstine M, Sannuti A, Molleston JP, et al. (octobre 2005). “Transplantation simultanée de foie et de pancréas chez des patients atteints de mucoviscidose”. Procédure de transplantation . 37 (8) : 3567–9. doi : 10.1016/j.transproceed.2005.09.091 . PMID 16298663 .
- ^ Belkin RA, Henig NR, Chanteur LG, Chaparro C, Rubenstein RC, Xie SX, et al. (mars 2006). “Facteurs de risque de décès de patients atteints de mucoviscidose en attente d’une transplantation pulmonaire” . Journal américain de médecine respiratoire et de soins intensifs . 173 (6): 659-66. doi : 10.1164/rccm.200410-1369OC . PMC 2662949 . PMID 16387803 .
- ^ un b “la Mucoviscidose – la Pédiatrie” . Édition professionnelle des manuels Merck . Récupéré le 12 août 2020 .
- ^ Somaraju UR, Solis-Moya A (août 2020). “Thérapie de remplacement des enzymes pancréatiques pour les personnes atteintes de fibrose kystique” . La base de données Cochrane des revues systématiques . 8 (9) : CD008227. doi : 10.1002/14651858.CD008227.pub4 . PMC 8094413 . PMID 32761612 .
- ^ Skolnik K, Levy RD, Wilcox PG, Quon BS (novembre 2016). “Maladie coronarienne dans la mucoviscidose : une préoccupation émergente ?” . Journal de la fibrose kystique . 15 (6) : e70–e71. doi : 10.1016/j.jcf.2016.09.010 . PMID 27751792 .
- ^ Zirbes J, Milla CE (septembre 2009). “Diabète lié à la fibrose kystique”. Examens respiratoires pédiatriques . 10 (3) : 118–23, quiz 123. doi : 10.1016/j.prrv.2009.04.004 . PMID 19651382 .
- ^ Onady GM, Stolfi A (octobre 2020). “Traitements médicamenteux pour la gestion du diabète lié à la fibrose kystique” . La base de données Cochrane des revues systématiques . 2020 (10) : CD004730. doi : 10.1002/14651858.CD004730.pub5 . PMC 8094754 . PMID 33075159 .
- ^ Amin R, Jahnke N, Waters V (mars 2020). “Traitement antibiotique pour Stenotrophomonas maltophilia chez les personnes atteintes de mucoviscidose” . La base de données Cochrane des revues systématiques . 3 : CD009249. doi : 10.1002/14651858.CD009249.pub5 . PMC 7080526 . PMID 32189337 .
- ^ un bc Conwell LS, Chang AB (mars 2014). “Bisphosphonates pour l’ostéoporose chez les personnes atteintes de mucoviscidose” . La base de données Cochrane des revues systématiques (3) : CD002010. doi : 10.1002/14651858.CD002010.pub4 . PMC 6718208 . PMID 24627308 .
- ^ Hardin DS, Rice J, Ahn C, Ferkol T, Howenstine M, Spears S, et al. (mars 2005). “Le traitement par l’hormone de croissance améliore la nutrition et la croissance chez les enfants atteints de mucoviscidose recevant une nutrition entérale”. Le Journal de pédiatrie . 146 (3): 324–8. doi : 10.1016/j.jpeds.2004.10.037 . PMID 15756212 .
- ^ Marques SC, Kissner DG (1997). “Gestion de la sinusite dans la fibrose kystique adulte”. Journal américain de rhinologie . 11 (1) : 11–4. doi : 10.2500/105065897781446810 . PMID 9065342 . S2CID 5606258 .
- ^ Phillipson GT, Petrucco OM, Matthews CD (février 2000). “Absence bilatérale congénitale du canal déférent, analyse des mutations de la fibrose kystique et injection intracytoplasmique de spermatozoïdes” . Reproduction humaine . 15 (2): 431–5. doi : 10.1093/humrep/15.2.431 . PMID 10655317 .
- ^ Ciofu O, Lykkesfeldt J (août 2014). “Supplémentation antioxydante pour les maladies pulmonaires dans la mucoviscidose” . La base de données Cochrane des revues systématiques . 8 (8) : CD007020. doi : 10.1002/14651858.CD007020.pub3 . PMC 6777741 . PMID 25102015 .
- ^ un b Radtke T, Nevitt SJ, Hebestreit H, Kriemler S (novembre 2017). “Entraînement à l’exercice physique pour la mucoviscidose” . La base de données Cochrane des revues systématiques . 2017 (11) : CD002768. doi : 10.1002/14651858.CD002768.pub4 . PMC 6485991 . PMID 29090734 .
- ^ Ganesan P, Schmiedge J, Manchaiah V, Swapna S, Dhandayutham S, Kothandaraman PP (avril 2018). « Ototoxicité : un défi dans le diagnostic et le traitement » . Journal d’audiologie et d’otologie . 22 (2): 59–68. doi : 10.7874/jao.2017.00360 . PMC 5894487 . PMID 29471610 .
- ^ Green J, Gilchrist FJ, Carroll W (juin 2018). “Interventions pour prévenir le syndrome d’obstruction intestinale distale (DIOS) dans la mucoviscidose” . La base de données Cochrane des revues systématiques . 6 : CD012619. doi : 10.1002/14651858.cd012619 . PMC 6478257 . PMID 29894558 .
- ^ Davis PB (mars 2006). “Mucoviscidose depuis 1938”. Journal américain de médecine respiratoire et de soins intensifs . 173 (5): 475–82. doi : 10.1164/rccm.200505-840OE . PMID 16126935 . S2CID 1770759 .
- ^ MacKenzie T, Gifford AH, Sabadosa KA, Quinton HB, Knapp EA, Goss CH, Marshall BC (août 2014). “Longévité des patients atteints de mucoviscidose de 2000 à 2010 et au-delà : analyse de la survie du registre des patients de la Cystic Fibrosis Foundation” . Annales de médecine interne . 161 (4): 233–241. doi : 10.7326/m13-0636 . PMC 4687404 . PMID 25133359 .
- ^ “Rapport du Registre canadien des données sur les patients atteints de fibrose kystique” (PDF) . Fondation canadienne de la fibrose kystique . 2007. Archivé de l’original (PDF) le 15 juillet 2010 . Récupéré le 14 mars 2010 .
- ^ “Rapport de données annuel 2016 Registre des patients de la Fondation de la fibrose kystique” (PDF) . p. 4. Archivé de l’original (PDF) le 19 juin 2018 . Récupéré le 19 juin 2018 .
- ^ “Rapport annuel de données du registre des patients atteints de fibrose kystique 2009” (PDF) . Fondation de la fibrose kystique . 2009. Archivé de l’original (PDF) le 5 janvier 2012.
- ^ Yu H, Nasr SZ, Deretic V (avril 2000). “Défenses pulmonaires innées et clairance compromise de Pseudomonas aeruginosa dans le modèle de souris mal nourrie d’infections respiratoires dans la mucoviscidose” . Infection et immunité . 68 (4): 2142–7. doi : 10.1128/IAI.68.4.2142-2147.2000 . PMC 97396 . PMID 10722612 .
- ^ Ratjen F, Döring G (février 2003). “Fibrose kystique”. Lancette . 361 (9358): 681–9. doi : 10.1016/S0140-6736(03)12567-6 . PMID 12606185 . S2CID 24879334 .
- ^ Rosenstein BJ, Zeitlin PL (janvier 1998). “Fibrose kystique”. Lancette . 351 (9098): 277–82. doi : 10.1016/S0140-6736(97)09174-5 . PMID 9457113 . S2CID 44627706 .
- ^ Schmitz TG, Goldbeck L (février 2006). « L’effet des programmes de réadaptation pour patients hospitalisés sur la qualité de vie des patients atteints de mucoviscidose : une étude multicentrique » . Résultats en matière de santé et de qualité de vie . 4 : 8. doi : 10.1186/1477-7525-4-8 . PMC 1373610 . PMID 16457728 .
- ^ Hegarty M, Macdonald J, Watter P, Wilson C (juillet 2009). “Qualité de vie des jeunes atteints de mucoviscidose : effets de l’hospitalisation, de l’âge et du sexe, et différences de perception parent/enfant”. Enfant . 35 (4) : 462–8. doi : 10.1111/j.1365-2214.2008.00900.x . PMID 18991968 .
Havermans T, Vreys M, Proesmans M, De Boeck C (janvier 2006). “Évaluation de l’accord entre parents et enfants sur la qualité de vie liée à la santé chez les enfants atteints de mucoviscidose”. Enfant . 32 (1): 1–7. doi : 10.1111/j.1365-2214.2006.00564.x . PMID 16398786 . - ^ Moorcroft AJ, Dodd ME, Webb AK (1998). “Limitations d’exercice et formation pour les patients atteints de fibrose kystique”. Handicap et réadaptation . 20 (6–7) : 247–253. doi : 10.3109/09638289809166735 . PMID 9637933 .
- ^ “Médicaments” . Fibrose kystique Canada. 2011. N° 10684-5100 RR0001. Archivé de l’original le 4 septembre 2011.
- ^ Araújo FG, Novaes FC, Santos NP, Martins VC, Souza SM, Santos SE, Ribeiro-dos-Santos AK (janvier 2005). “Prévalence des mutations deltaF508, G551D, G542X et R553X chez les patients atteints de mucoviscidose dans le nord du Brésil” . Journal brésilien de recherche médicale et biologique . 38 (1) : 11–5. doi : 10.1590/S0100-879X2005000100003 . PMID 15665983 .
- ^ Tobias E (2011). Génétique médicale essentielle . John Wiley et fils. p. 312. ISBN 978-1-118-29370-6. Archivé de l’original le 17 avril 2016.
- ^ “Les faits et chiffres canadiens sur la fibrose kystique” . fibrosekystique.ca . Archivé de l’original le 16 juin 2013.
- ^ “Test de Porteur Génétique” . Fondation de la fibrose kystique. 2007. Archivé de l’original le 23 mars 2010.
- ^ Rosenstein BJ, Couper GR (avril 1998). “Le diagnostic de fibrose kystique : une déclaration de consensus. Panneau de consensus de la Fondation de la fibrose kystique”. Le Journal de pédiatrie . 132 (4): 589–95. doi : 10.1016/S0022-3476(98)70344-0 . PMID 9580754 .
- ^ Hamosh A, FitzSimmons SC, Macek M, Knowles MR, Rosenstein BJ, Cutting GR (février 1998). “Comparaison des manifestations cliniques de la mucoviscidose chez les patients noirs et blancs”. Le Journal de pédiatrie . 132 (2) : 255–9. doi : 10.1016/S0022-3476(98)70441-X . PMID 9506637 .
- ^ Farrell P, Joffe S, Foley L, Canny GJ, Mayne P, Rosenberg M (septembre 2007). « Diagnostic de la mucoviscidose en République d’Irlande : épidémiologie et coûts » . Journal médical irlandais . 100 (8) : 557–60. PMID 17955689 . Archivé de l’original le 3 décembre 2013.
- ^ Hytönen M, Patjas M, Vento SI, Kauppi P, Malmberg H, Ylikoski J, Kere J (décembre 2001). “Mutations du gène de la fibrose kystique deltaF508 et 394delTT chez les patients atteints de sinusite chronique en Finlande”. Acta Oto-Laryngologique . 121 (8) : 945–7. doi : 10.1080/000164801317166835 . PMID 11813900 .
- ^ “QUI | Gènes et maladie humaine” . Qui.int. 7 décembre 2010. Archivé de l’original le 20 octobre 2012 . Récupéré le 23 janvier 2013 .
- ^ Russel P (2011). Biologie: la science dynamique (2e éd.). Belmont, Californie : Brooks/Cole, Cengage Learning. p. 304.ISBN _ 978-0-538-49372-7. Archivé de l’original le 17 avril 2016.
- ^ “Tests génétiques pour la fibrose kystique Tests génétiques pour la fibrose kystique” . Déclaration de la conférence de développement de consensus . Instituts nationaux de la santé. 14-16 avril 1997. Archivé de l’original le 27 mars 2009.
- ^ Rosenfeld M, Davis R, FitzSimmons S, Pepe M, Ramsey B (mai 1997). “L’écart entre les sexes dans la mortalité de la fibrose kystique” . Journal américain d’épidémiologie . 145 (9): 794–803. doi : 10.1093/oxfordjournals.aje.a009172 . PMID 9143209 .
- ^ Coakley RD, Sun H, Clunes LA, Rasmussen JE, Stackhouse JR, Okada SF, et al. (décembre 2008). “17beta-Estradiol inhibe l’homéostasie dépendante du Ca2+ du volume de liquide de surface des voies respiratoires dans l’épithélium des voies respiratoires de la fibrose kystique humaine” . Le Journal d’investigation clinique . 118 (12): 4025–35. doi : 10.1172/JCI33893 . PMC 2582929 . PMID 19033671 .
- ^ Verma N, Bush A, Buchdahl R (octobre 2005). « Y a-t-il encore un écart entre les sexes dans la mucoviscidose ? » Poitrine . 128 (4): 2824–34. doi : 10.1378/chest.128.4.2824 . PMID 16236961 .
- ^ Moran A, Dunitz J, Nathan B, Saeed A, Holme B, Thomas W (septembre 2009). “Diabète lié à la fibrose kystique : tendances actuelles de la prévalence, de l’incidence et de la mortalité” . Soins du diabète . 32 (9) : 1626–1631. doi : 10.2337/dc09-0586 . PMC 2732133 . PMID 19542209 .
- ^ “CF pire pour les femmes” en raison de l’effet des œstrogènes ” ” . The Irish Times . 8 août 2010. Archivé de l’original le 11 août 2010.
- ^ Wennberg C, Kucinskas V (1994). “Faible fréquence de la mutation delta F508 dans les populations finno-ougriennes et baltes”. Hérédité humaine . 44 (3): 169–71. doi : 10.1159/000154210 . PMID 8039801 .
- ^ Kere J, Savilahti E, Norio R, Estivill X, de la Chapelle A (septembre 1990). « Mutation delta F508 de mucoviscidose en Finlande : d’autres mutations prédominent ». Génétique humaine . 85 (4) : 413–5. doi : 10.1007/BF02428286 . PMID 2210753 . S2CID 38364780 .
- ^ Wiuf C (août 2001). “Les hétérozygotes delta F508 ont-ils un avantage sélectif?”. Recherche génétique . 78 (1): 41–7. CiteSeerX 10.1.1.174.7283 . doi : 10.1017/S0016672301005195 . PMID 11556136 .
- ^ Gabriel SE, Brigman KN, Koller BH, Boucher RC, Stutts MJ (octobre 1994). “Résistance hétérozygote de la fibrose kystique à la toxine du choléra dans le modèle murin de la fibrose kystique”. Sciences . 266 (5182): 107–9. Bib code : 1994Sci …266..107G . doi : 10.1126/science.7524148 . PMID 7524148 .
- ^ Alfonso-Sánchez MA, Pérez-Miranda AM, García-Obregón S, Peña JA (juin 2010). “Une approche évolutive de la fréquence élevée de la mutation Delta F508 CFTR dans les populations européennes”. Hypothèses médicales . 74 (6): 989–92. doi : 10.1016/j.mehy.2009.12.018 . PMID 20110149 .
- ^ Cuthbert AW, Halstead J, Ratcliff R, Colledge WH, Evans MJ (janvier 1995). “L’hypothèse de l’avantage génétique chez les hétérozygotes de la mucoviscidose : une étude murine” . Le Journal de Physiologie . 482 (Pt 2): 449–54. doi : 10.1113/jphysiol.1995.sp020531 . PMC 1157742 . PMID 7714835 .
- ^ Högenauer C, Santa Ana CA, Porter JL, Millard M, Gelfand A, Rosenblatt RL, et al. (Décembre 2000). “Sécrétion intestinale active de chlorure chez les porteurs humains de mutations de la fibrose kystique : une évaluation de l’hypothèse selon laquelle les hétérozygotes ont une sécrétion intestinale active de chlorure sous-normale” . Journal américain de génétique humaine . 67 (6) : 1422–7. doi : 10.1086/316911 . PMC 1287919 . PMID 11055897 .
- ^ Pier GB, Grout M, Zaidi T, Meluleni G, Mueschenborn SS, Banting G, et al. (mai 1998). “Salmonella typhi utilise le CFTR pour pénétrer dans les cellules épithéliales intestinales”. Nature . 393 (6680): 79–82. Bibcode : 1998Natur.393…79P . doi : 10.1038/30006 . PMID 9590693 . S2CID 5894247 .
- ^ Modiano G, Ciminelli BM, Pignatti PF (mars 2007). « Fibrose kystique et persistance de la lactase : une corrélation possible ». Journal européen de génétique humaine . 15 (3): 255–9. doi : 10.1038/sj.ejhg.5201749 . PMID 17180122 . S2CID 4650571 .
- ^ Poolman EM, Galvani AP (février 2007). “Évaluer les candidats agents de pression sélective pour la mucoviscidose” . Journal de la Royal Society, Interface . 4 (12): 91–8. doi : 10.1098/rsif.2006.0154 . PMC 2358959 . PMID 17015291 .
- ^ Williams N (2006). “L’empreinte craint une nouvelle menace de tuberculose” . Biologie actuelle . 16 (19) : R821–R822. doi : 10.1016/j.cub.2006.09.009 . S2CID 2346727 .
- ^ Tobacman JK (juin 2003). “La carence en arylsulfatase B joue-t-elle un rôle dans la mucoviscidose ?”. Poitrine . 123 (6): 2130–9. doi : 10.1378/chest.123.6.2130 . PMID 12796199 .
- ^ un bc Busch R (1990). “Sur l’histoire de la fibrose kystique”. Acta Universitatis Carolinae. Médicament . 36 (1–4) : 13–15. PMID 2130674 .
- ^ Fanconi G, Uehlinger E, Knauer C (1936). “Das coeliakiesyndrom bei angeborener zysticher pankreasfibromatose und bronchiektasien”. Vienne. Méd. WSCHR . 86 : 753–756.
- ^ Di Sant’Agnese PA, Darling RC, Perera GA, Shea E (novembre 1953). “Composition électrolytique anormale de la sueur dans la fibrose kystique du pancréas; signification clinique et relation avec la maladie”. Pédiatrie . 12 (5): 549–563. doi : 10.1542/peds.12.5.549 . PMID 13111855 . S2CID 42514224 .
- ^ Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. (septembre 1989). “Identification du gène de la mucoviscidose : clonage et caractérisation de l’ADN complémentaire”. Sciences . 245 (4922): 1066–1073. Bibcode : 1989Sci…245.1066R . doi : 10.1126/science.2475911 . PMID 2475911 .
- ^ Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, et al. (septembre 1989). “Identification du gène de la mucoviscidose : marche et saut des chromosomes”. Sciences . 245 (4922): 1059-1065. Bibcode : 1989Sci…245.1059R . doi : 10.1126/science.2772657 . PMID 2772657 .
- ^ Freudenheim M (22 décembre 2009). “Outil dans la lutte contre la fibrose kystique : un registre” . Le New York Times . p. D1. Archivé de l’original le 24 mai 2013 . Récupéré le 21 décembre 2009 .
- ^ Lee TW, Southern KW, Perry LA, Penny-Dimri JC, Aslam AA (juin 2016). Southern KW (éd.). “Remplacement topique du gène régulateur de la conductance transmembranaire de la fibrose kystique pour les maladies pulmonaires liées à la fibrose kystique” . La base de données Cochrane des revues systématiques . 2016 (6) : CD005599. doi : 10.1002/14651858.CD005599.pub5 . PMC 8682957 . PMID 27314455 .
- ^ Alton EW, Armstrong DK, Ashby D, Bayfield KJ, Bilton D, Bloomfield EV, et al. (septembre 2015). “Nébulisation répétée de la thérapie génique CFTR non virale chez les patients atteints de mucoviscidose : un essai de phase 2b randomisé, en double aveugle, contrôlé par placebo” . Le Lancet. Médecine Respiratoire . 3 (9): 684–691. doi : 10.1016/S2213-2600(15)00245-3 . PMC 4673100 . PMID 26149841 .
- ^ Ramalho AS, Beck S, Meyer M, Penque D, Cutting GR, Amaral MD (novembre 2002). “Cinq pour cent de l’ARNm régulateur de la conductance transmembranaire de la fibrose kystique normale améliore la gravité de la maladie pulmonaire dans la fibrose kystique”. Journal américain de biologie cellulaire et moléculaire respiratoire . 27 (5) : 619–627. doi : 10.1165/rcmb.2001-0004oc . PMID 12397022 . S2CID 8714332 .
- ^ Tate S, Elborn S (mars 2005). “Progrès vers la thérapie génique pour la mucoviscidose”. Avis d’expert sur l’administration de médicaments . 2 (2): 269–80. doi : 10.1517/17425247.2.2.269 . PMID 16296753 . S2CID 30948229 .
- ^ Héritage mendélien en ligne chez l’homme (OMIM): FIBROSE KYSTIQUE ; CF-219700
- ^ Schwank G, Koo BK, Sasselli V, Dekkers JF, Heo I, Demircan T, et al. (Décembre 2013). “Réparation fonctionnelle de CFTR par CRISPR/Cas9 dans les organoïdes de cellules souches intestinales de patients atteints de mucoviscidose” . Cellule Cellule Souches . 13 (6): 653–8. doi : 10.1016/j.stem.2013.11.002 . PMID 24315439 .
- ^ Hraiech S, Brégeon F, Rolain JM (2015). “Traitement à base de bactériophages dans les infections à Pseudomonas aeruginosa associées à la fibrose kystique : justification et état actuel” . Conception, développement et thérapie de médicaments . 9 : 3653–63. doi : 10.2147/DDDT.S53123 . PMC 4509528 . PMID 26213462 .
- ^ Tendance S, Fonceca AM, Ditcham WG, Kicic A, Cf A (novembre 2017). “Le potentiel de la thérapie par les phages dans la mucoviscidose : interactions essentielles entre les bactéries et les phages humains et considérations d’administration à utiliser dans les voies respiratoires infectées par Pseudomonas aeruginosa” . Journal de la fibrose kystique . 16 (6): 663–670. doi : 10.1016/j.jcf.2017.06.012 . PMID 28720345 .
- ^ un b Ramsey BW, Downey GP, Goss CH (mai 2019). “Mise à jour sur la fibrose kystique 2018” . Journal américain de médecine respiratoire et de soins intensifs . 199 (10): 1188–1194. doi : 10.1164/rccm.201902-0310UP . PMC 6519861 . PMID 30917288 . ProQuest 2230820891 .
- ^ Dietz HC (août 2010). “Nouvelles approches thérapeutiques des troubles mendéliens”. Le New England Journal of Medicine . 363 (9) : 852–863. doi : 10.1056/NEJMra0907180 . PMID 20818846 . S2CID 5809127 . Texte intégral gratuit
- ^ a b Bureau du commissaire (24 octobre 2019). “La FDA approuve une nouvelle thérapie révolutionnaire pour la mucoviscidose” . FDA . Récupéré le 13 novembre 2019 .
- ^ “Thérapies de modulateur CFTR” . Bethesda, Md. : Fondation de la fibrose kystique . Récupéré le 13 novembre 2019 .
- ^ Sherrard LJ, McGrath SJ, McIlreavey L, Hatch J, Wolfgang MC, Muhlebach MS, Gilpin DF, Elborn JS, Tunney MM (février 2016). “Production de bêta-lactamases à spectre étendu et rôle pathogène indirect potentiel des isolats de Prevotella à partir du microbiote respiratoire de la mucoviscidose” . Int J Agents antimicrobiens . 47 (2): 140–145. doi : 10.1016/j.ijantimicag.2015.12.004 . PMC 4746055 . PMID 26774156 .
Ouvrages cités
- Egan ME, Schechter MS, Voynow JA (2020). “Fibrose kystique”. Dans Kliegman RM, St Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds.). Manuel Nelson de pédiatrie . Elsevier. pages 2282–2297. ISBN 978-0-323-56890-6.
Liens externes
| Wikimedia Commons a des médias liés à la fibrose kystique . |
- Recherchez sur GeneCards les gènes impliqués dans la mucoviscidose
- Base de données sur les mutations de la fibrose kystique
- « Fibrose kystique » . MedlinePlus . Bibliothèque nationale de médecine des États-Unis.