Alcène
En chimie organique , un alcène est un hydrocarbure contenant une double liaison carbone -carbone . [1]
 Un modèle 3D de l’éthylène , l’alcène le plus simple.
Un modèle 3D de l’éthylène , l’alcène le plus simple.
Alcène est souvent utilisé comme synonyme d’ oléfine , c’est-à-dire tout hydrocarbure contenant une ou plusieurs doubles liaisons. [2] On distingue deux types généraux de monoalcènes : terminal et interne. Aussi appelés α-oléfines , les alcènes terminaux sont plus utiles.
Cependant, l ‘ Union internationale de chimie pure et appliquée (IUPAC) recommande d’utiliser le nom «alcène» uniquement pour les hydrocarbures acycliques avec une seule double liaison; alcadiène , alcatriène , etc., ou polyène pour les hydrocarbures acycliques avec deux ou plusieurs doubles liaisons; cycloalcène , cycloalcadiène , etc. pour les cycliques ; et “oléfine” pour la classe générale – cyclique ou acyclique, avec une ou plusieurs doubles liaisons. [3] [4] [5]
Les alcènes acycliques, avec une seule double liaison et aucun autre groupe fonctionnel (également appelés mono-ènes ) forment une série homologue d’ hydrocarbures avec la formule générale C n H 2 n avec n étant 2 ou plus (ce qui correspond à deux hydrogènes de moins que le alcane correspondant ). Lorsque n vaut quatre ou plus, des isomères sont possibles, se distinguant par la position et la conformation de la double liaison.
Les alcènes sont généralement des composés non polaires incolores , quelque peu similaires aux alcanes mais plus réactifs. Les premiers membres de la série sont des gaz ou des liquides à température ambiante. L’alcène le plus simple, l’éthylène ( C 2 H 4 ) (ou « éthène » dans la nomenclature IUPAC ) est le composé organique produit à plus grande échelle industriellement. [6]
Les composés aromatiques sont souvent dessinés comme des alcènes cycliques, mais leur structure et leurs propriétés sont suffisamment distinctes pour qu’ils ne soient pas classés comme alcènes ou oléfines. [4] Les hydrocarbures avec deux doubles liaisons superposées ( C=C=C ) sont appelés allènes – le composé le plus simple est lui-même appelé allène – et ceux avec trois liaisons superposées ou plus ( C=C=C=C , C=C= C=C=C , etc.) sont appelés cumulènes . Certains auteurs [ qui ? ] ne considèrent pas les allènes et les cumulènes comme des “alcènes”.
Isomérie structurale
Les alcènes ayant quatre atomes de carbone ou plus peuvent former divers isomères structuraux . La plupart des alcènes sont également des isomères de cycloalcanes . Les isomères structuraux d’alcènes acycliques avec une seule double liaison suivent: [7]
- C 2 H 4 : éthylène seul
- C 3 H 6 : Propylène uniquement
- C 4 H 8 : 3 isomères : 1-butène , 2-butène , et isobutylène
- C 5 H 10 : 5 isomères : 1-pentène , 2-pentène, 2-méthyl-1-butène, 3-méthyl-1-butène, 2-méthyl-2-butène
- C 6 H 12 : 13 isomères :
1-hexène, cis-2-hexène, trans-2-hexène, cis-3-hexène, trans-3-hexène, 2 méthyl 1-pentène, 3 méthyl 1-pentène, 4 méthyl 1-pentène, 2 méthyl 2- pentène, 3 méthyl 2-pentène, 4 méthyl 2-pentène, 2 3 diméthyl 1-butène, 2 3 diméthyl 2-butène
- C 7 H 14 : 27 isomères (calculés)
- C 12 H 24 : 2 281 isomères (calculés)
- C 31 H 62 : 193 706 542 776 isomères (calculés)
Beaucoup de ces molécules présentent une isomérie cis-trans . Il peut également y avoir des atomes de carbone chiraux , en particulier dans les molécules plus grosses (à partir de C 5 ). Le nombre d’isomères potentiels augmente rapidement avec des atomes de carbone supplémentaires.
Structure et collage
Collage
 Éthylène (éthène), montrant la liaison pi en vert
Éthylène (éthène), montrant la liaison pi en vert
Une double liaison carbone-carbone est constituée d’une liaison sigma et d’une liaison pi . Cette double liaison est plus forte qu’une simple liaison covalente (611 kJ / mol pour C=C contre 347 kJ/mol pour C–C), [1] mais pas deux fois plus forte. Les doubles liaisons sont plus courtes que les liaisons simples avec une longueur de liaison moyenne de 1,33 Å (133 pm ) contre 1,53 Å pour une simple liaison CC typique. [8]
Chaque atome de carbone de la double liaison utilise ses trois orbitales hybrides sp 2 pour former des liaisons sigma à trois atomes (l’autre atome de carbone et deux atomes d’hydrogène). Les orbitales atomiques 2p non hybridées, qui sont perpendiculaires au plan créé par les axes des trois orbitales hybrides sp2, se combinent pour former la liaison pi. Cette liaison se situe à l’extérieur de l’axe principal C – C, avec la moitié de la liaison d’un côté de la molécule et l’autre moitié de l’autre. Avec une force de 65 kcal/mol, la liaison pi est significativement plus faible que la liaison sigma.
La rotation autour de la double liaison carbone-carbone est limitée car elle entraîne un coût énergétique pour rompre l’alignement des orbitales p sur les deux atomes de carbone. Par conséquent , les isomères cis ou trans s’interconvertissent si lentement qu’ils peuvent être manipulés librement dans des conditions ambiantes sans isomérisation. Des alcènes plus complexes peuvent être nommés avec la notation E – Z pour les molécules avec trois ou quatre substituants différents (groupes latéraux). Par exemple, parmi les isomères du butène , les deux groupes méthyle du ( Z )-but-2 -ène (alias cis –2-butène) apparaissent du même côté de la double liaison, et dans (E )-but-2-ène (alias trans –2-butène) les groupes méthyle apparaissent sur les côtés opposés. Ces deux isomères du butène ont des propriétés distinctes.
Façonner
Comme prédit par le modèle VSEPR de répulsion des paires d’ électrons , la géométrie moléculaire des alcènes comprend des angles de liaison autour de chaque atome de carbone dans une double liaison d’environ 120°. L’angle peut varier en raison de la contrainte stérique introduite par les Interactions non liées entre les groupes fonctionnels attachés aux atomes de carbone de la double liaison. Par exemple, l’Angle de liaison C – C – C dans le Propylène est de 123,9 °.
Pour les alcènes pontés, la règle de Bredt stipule qu’une double liaison ne peut pas se produire à la tête de pont d’un système d’anneaux pontés à moins que les anneaux ne soient suffisamment grands. [9] Suivant Fawcett et définissant S comme le nombre total d’atomes non têtes de pont dans les anneaux, [10] les systèmes bicycliques nécessitent S ≥ 7 pour la stabilité [9] et les systèmes tricycliques nécessitent S ≥ 11. [11]
Propriétés physiques
De nombreuses propriétés physiques des alcènes et des alcanes sont similaires : ils sont incolores, non polaires et combustibles. L’ État physique dépend de la masse moléculaire : comme les hydrocarbures saturés correspondants, les alcènes les plus simples ( éthylène , Propylène et butène ) sont des gaz à température ambiante. Les alcènes linéaires d’environ cinq à seize atomes de carbone sont des liquides et les alcènes supérieurs sont des solides cireux. Le point de fusion des solides augmente également avec l’augmentation de la masse moléculaire.
Les alcènes ont généralement des odeurs plus fortes que leurs alcanes correspondants. L’éthylène a une odeur sucrée et de moisi. La liaison de l’ion cuivrique à l’oléfine dans le récepteur olfactif des mammifères MOR244-3 est impliquée dans l’odeur des alcènes (ainsi que des thiols). Les alcènes tendus, en particulier, comme le norbornène et le trans – cyclooctène sont connus pour avoir des odeurs fortes et désagréables, un fait cohérent avec les complexes π plus forts qu’ils forment avec les ions métalliques, y compris le cuivre. [12]
Réactions
Les alcènes sont des composés relativement stables, mais ils sont plus réactifs que les alcanes . La plupart des réactions des alcènes impliquent des ajouts à cette liaison pi, formant de nouvelles liaisons simples . Les alcènes servent de matière première pour l’ industrie pétrochimique car ils peuvent participer à une grande variété de réactions, principalement la polymérisation et l’alkylation.
À l’exception de l’éthylène, les alcènes ont deux sites de réactivité : la liaison carbone-carbone pi et la présence de centres CH allyliques . Les premiers dominent mais les sites allyliques sont importants aussi.
Réactions d’addition
Les alcènes réagissent dans de nombreuses réactions d’addition , qui se produisent en ouvrant la double liaison. La plupart de ces réactions d’addition suivent le mécanisme de l’addition électrophile . Des exemples sont l’ hydrohalogénation , l’halogénation , la Formation d’halohydrine , l’ Oxymercuration , l’ hydroboration , l’addition de dichlorocarbène , la réaction de Simmons-Smith , l’Hydrogénation catalytique , l’ Époxydation , la polymérisation radicalaire et l’ hydroxylation .
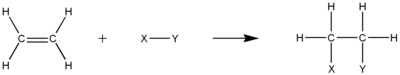 Hydrogénation et hydroéléments associés
Hydrogénation et hydroéléments associés
L’hydrogénation des alcènes produit les alcanes correspondants . La réaction est parfois effectuée sous pression et à température élevée. Les catalyseurs métalliques sont presque toujours nécessaires. Les catalyseurs industriels courants sont à base de platine , de nickel et de palladium . Une application à grande échelle est la production de margarine .
Outre l’ajout de HH à travers la double liaison, de nombreux autres HX peuvent être ajoutés. Ces procédés ont souvent une grande importance commerciale. Un exemple est l’addition de H-SiR3 , c’est-à-dire l’ hydrosilylation . Cette réaction est utilisée pour générer des composés organosiliciés . Une autre réaction est l’ hydrocyanation , l’addition de H-CN à travers la double liaison.
Hydratation
L’ hydratation , l’ajout d’eau à travers la double liaison des alcènes, donne des alcools . La réaction est catalysée par l’acide phosphorique ou l’acide sulfurique . Cette réaction est réalisée à l’échelle industrielle pour produire de l’ éthanol de synthèse .
CH 2 =CH 2 + H 2 O → CH 3 -CH 2 OH
Les alcènes peuvent également être convertis en alcools via la Réaction d’oxymercuration-démercuration , la réaction d’hydroboration-oxydation ou par hydratation de Mukaiyama .
Halogénation
Dans l’halogénation électrophile, l’addition de brome ou de chlore élémentaire aux alcènes donne respectivement des dibromo- et dichloroalcanes vicinaux (1,2-dihalogénures ou dihalogénures d’éthylène). La décoloration d’une solution de brome dans l’eau est un test analytique de la présence d’alcènes :
CH 2 =CH 2 + Br 2 → BrCH 2 –CH 2 Br
Les réactions apparentées sont également utilisées comme mesures quantitatives de l’insaturation, exprimées par l’ indice de brome et l’indice d’ iode d’un composé ou d’un mélange.
Hydrohalogénation
L’hydrohalogénation est l’addition d’halogénures d’hydrogène , tels que HCl ou HI , à des alcènes pour donner les haloalcanes correspondants :
CH 3 -CH=CH 2 + HI → CH 3 -CH I -CH 2 – H
Si les deux atomes de carbone de la double liaison sont liés à un nombre différent d’atomes d’hydrogène, l’halogène se trouve préférentiellement sur le carbone avec moins de substituants hydrogène. Ce modèle est connu sous le nom de règle de Markovnikov . L’utilisation d’ initiateurs radicalaires ou d’autres composés peut conduire au résultat de produit opposé. L’acide bromhydrique en particulier est susceptible de former des radicaux en présence de diverses impuretés ou même d’oxygène atmosphérique, conduisant à l’inversion du résultat de Markovnikov : [13]
CH 3 -CH=CH 2 + HBr → CH 3 -CH H -CH 2 – Br Formation d’halohydrine
Les alcènes réagissent avec l’eau et les halogènes pour former des halohydrines par une réaction d’addition. La régiochimie et l’anti-stéréochimie de Markovnikov se produisent.
CH 2 =CH 2 + X 2 + H 2 O → XCH 2 -CH 2 OH + HX Oxydation
Les alcènes réagissent avec les acides percarboxyliques et même le peroxyde d’hydrogène pour donner des époxydes :
RCH = CH 2 + RCO 3 H → RCHOCH 2 + RCO 2 H
Pour l’éthylène, l’ Époxydation est réalisée à très grande échelle industriellement à l’aide d’oxygène en présence de catalyseurs :
C 2 H 4 + 1/2 O 2 → C 2 H 4 O
Les alcènes réagissent avec l’ozone, entraînant la scission de la double liaison. Le processus s’appelle l’ ozonolyse . Souvent, la procédure de réaction comprend un réducteur doux, tel que le sulfure de diméthyle (SMe 2 ):
RCH=CHR’ + O 3 + SMe 2 → RCHO + R’CHO + O=SMe 2 R 2 C=CHR’ + O 3 → R 2 CHO + R’CHO + O=SMe 2
Lorsqu’ils sont traités avec une solution chaude concentrée et acidifiée de KMnO 4 , les alcènes sont des cétones et/ou des acides carboxyliques clivés . La stoechiométrie de la réaction est sensible aux conditions. Cette réaction et l’ozonolyse peuvent être utilisées pour déterminer la position d’une double liaison dans un alcène inconnu.
L’oxydation peut être arrêtée au diol vicinal plutôt qu’au clivage complet de l’alcène en utilisant du tétroxyde d’osmium ou d’autres oxydants :
R’CH=CR 2 + 1/2 O 2 + H 2 O → R’CH(OH)-C(OH)R 2
Cette réaction est appelée dihydroxylation .
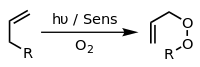
En présence d’un photosensibilisateur approprié , tel que le bleu de méthylène et la lumière, les alcènes peuvent subir une réaction avec les espèces réactives de l’oxygène générées par le photosensibilisateur, telles que les radicaux hydroxyles , l’oxygène singulet ou l’ion superoxyde . Les réactions du sensibilisateur excité peuvent impliquer un transfert d’électrons ou d’hydrogène, généralement avec un substrat réducteur (réaction de type I) ou une interaction avec l’oxygène (réaction de type II). [14] Ces divers processus et réactions alternatifs peuvent être contrôlés par le choix de conditions de réaction spécifiques, conduisant à une large gamme de produits. Un exemple courant est la [4+2] -cycloaddition d’oxygène singulet avec un diènetel que le cyclopentadiène pour donner un endoperoxyde :
![Generation of singlet oxygen and its [4+2]-cycloaddition with cyclopentadiene](http://upload.wikimedia.org/wikipedia/commons/thumb/1/12/4%2B2_cycloaddition_cyclopentadiene_O2.svg/350px-4%2B2_cycloaddition_cyclopentadiene_O2.svg.png)
Un autre exemple est la réaction ène de Schenck , dans laquelle l’oxygène singulet réagit avec une structure Allylique pour donner un peroxyde d’allyle transposé :
Polymérisation
Les alcènes terminaux sont des précurseurs de polymères via des processus appelés polymérisation . Certaines polymérisations ont une grande importance économique, car elles génèrent comme les plastiques polyéthylène et polypropylène . Les polymères d’alcène sont généralement appelés polyoléfines bien qu’ils ne contiennent pas d’oléfines. La polymérisation peut se dérouler via divers mécanismes. les diènes conjugués tels que le buta-1,3-diène et l’ isoprène (2-méthylbuta-1,3-diène) produisent également des polymères, un exemple étant le caoutchouc naturel.
Complexation des métaux
 Structure du bis (cyclooctadiène) nickel (0) , un complexe métal-alcène
Structure du bis (cyclooctadiène) nickel (0) , un complexe métal-alcène
Les alcènes sont des ligands dans les complexes d’alcènes de métaux de transition . Les deux centres de carbone se lient au métal à l’aide des orbitales C – C pi et pi *. Les mono- et dioléfines sont souvent utilisées comme ligands dans des complexes stables. Le cyclooctadiène et le norbornadiène sont des agents chélateurs populaires, et même l’éthylène lui-même est parfois utilisé comme ligand, par exemple dans le sel de Zeise . De plus, les complexes métal-alcène sont des intermédiaires dans de nombreuses réactions catalysées par un métal, notamment l’hydrogénation, l’hydroformylation et la polymérisation.
Aperçu de la réaction
| Nom de la réaction | Produit | Commenter |
|---|---|---|
| Hydrogénation | alcanes | ajout d’hydrogène |
| Hydroalcénylation | alcènes | hydrométallation / insertion / béta-élimination par Catalyseur métallique |
| Réaction d’addition d’halogène | 1,2-dihalogénure | addition électrophile d’halogènes |
| Hydrohalogénation ( Markovnikov ) | haloalcanes | addition d’acides halohydriques |
| Hydrohalogénation anti-Markovnikov | haloalcanes | addition médiée par les radicaux libres d’acides halohydriques |
| Hydroamination | amines | ajout d’une liaison N – H à travers la double liaison C – C |
| Hydroformylation | aldéhydes | procédé industriel, ajout de CO et H 2 |
| Hydrocarboxylation et réaction de Koch | acide carboxylique | procédé industriel, ajout de CO et H 2 O. |
| Carboalcoxylation | ester | procédé industriel, ajout de CO et d’alcool. |
| alkylation | ester | procédé industriel : alcène alkylant l’acide carboxylique avec l’acide silicotungstique comme Catalyseur. |
| Bishydroxylation sans netteté | diols | oxydation, réactif : tétroxyde d’osmium, ligand chiral |
| cis -hydroxylation de Woodward | diols | oxydation, réactifs : iode, acétate d’argent |
| Ozonolyse | aldéhydes ou cétones | réactif : ozone |
| Métathèse d’oléfine | alcènes | deux alcènes se réarrangent pour former deux nouveaux alcènes |
| Réaction de Diels-Alder | cyclohexènes | cycloaddition avec un diène |
| Réaction de Pauson-Khand | cyclopenténones | cycloaddition avec un alcyne et CO |
| Hydroboration–oxydation | alcools | réactifs : borane, puis un peroxyde |
| Oxymercuration-réduction | alcools | addition électrophile d’acétate mercurique, puis réduction |
| Réaction de Prins | 1,3-diols | addition électrophile avec un aldéhyde ou une cétone |
| Réaction de Paterno-Büchi | oxétanes | réaction photochimique avec un aldéhyde ou une cétone |
| Époxydation | époxyde | addition électrophile d’un peroxyde |
| Cyclopropanation | cyclopropanes | ajout de carbènes ou carbénoïdes |
| Hydroacylation | cétones | addition oxydante / élimination réductrice par Catalyseur métallique |
| Hydrophosphination | phosphines |
Synthèse
Méthodes industrielles
Les alcènes sont produits par craquage d’hydrocarbures . Les matières premières sont principalement des composants de condensat de gaz naturel (principalement de l’éthane et du propane) aux États-Unis et au Moyen-Orient et du naphta en Europe et en Asie. Les alcanes sont séparés à des températures élevées, souvent en présence d’un Catalyseur zéolite , pour produire un mélange d’alcènes principalement aliphatiques et d’alcanes de poids moléculaire inférieur. Le mélange dépend de la charge d’alimentation et de la température, et est séparé par distillation fractionnée. Ceci est principalement utilisé pour la fabrication de petits alcènes (jusqu’à six carbones). [15]
![]()
![]()
La déshydrogénation catalytique est liée à cela , où un alcane perd de l’hydrogène à des températures élevées pour produire un alcène correspondant. [1] C’est l’inverse de l’ Hydrogénation catalytique des alcènes.

Ce processus est également connu sous le nom de reformage . Les deux processus sont endothermiques et sont entraînés vers l’alcène à haute température par entropie .
La synthèse catalytique d’α-alcènes supérieurs (du type RCH=CH 2 ) peut également être réalisée par une réaction de l’éthylène avec le composé organométallique triéthylaluminium en présence de nickel , de cobalt ou de platine .
Réactions d’élimination
L’une des principales méthodes de synthèse d’alcènes en laboratoire est l’ élimination en salle des halogénures d’alkyle, des alcools et des composés similaires. La plus courante est la β-élimination via le mécanisme E2 ou E1, [16] mais les α-éliminations sont également connues.
Le mécanisme E2 fournit une méthode de β-élimination plus fiable que E1 pour la plupart des synthèses d’alcènes. La plupart des éliminations E2 commencent par un ester d’halogénure d’alkyle ou de sulfonate d’alkyle (tel qu’un tosylate ou un triflate ). Lorsqu’un halogénure d’alkyle est utilisé, la réaction est appelée déshydrohalogénation . Pour les produits asymétriques, les alcènes les plus substitués (ceux avec moins d’hydrogènes attachés au C = C) ont tendance à prédominer (voir la règle de Zaitsev ). Deux méthodes courantes de réactions d’élimination sont la déshydrohalogénation des halogénures d’alkyle et la déshydratation des alcools. Un exemple typique est illustré ci-dessous ; notez que si possible, le H est anti au groupe partant, même si cela conduit à l’ isomère Z le moins stable .[17]
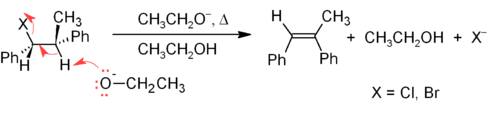
Les alcènes peuvent être synthétisés à partir d’alcools par déshydratation , auquel cas l’eau est perdue via le mécanisme E1. Par exemple, la déshydratation de l’ éthanol produit de l’éthylène :
CH 3 CH 2 OH → H 2 C=CH 2 + H 2 O
Un alcool peut également être converti en un meilleur groupe partant (par exemple, xanthate ), de manière à permettre une syn -élimination plus douce telle que l’ élimination de Chugaev et l’ élimination de Grieco . Les réactions apparentées comprennent les éliminations par les β-haloéthers (la synthèse des oléfines Boord ) et les esters ( pyrolyse des esters ).
Les alcènes peuvent être préparés indirectement à partir d’ alkylamines . L’amine ou l’ammoniac n’est pas un groupe partant approprié, de sorte que l’amine est d’abord soit alkylée (comme dans l’ élimination de Hofmann ) soit oxydée en un oxyde d’amine (la réaction de Cope ) pour rendre possible une élimination en douceur. La réaction Cope est une syn -élimination qui se produit à ou en dessous de 150 °C, par exemple : [18]

L’élimination de Hofmann est inhabituelle en ce que l’alcène le moins substitué (non Zaitsev ) est généralement le produit principal.
Les alcènes sont générés à partir d’α-halo sulfones dans la réaction de Ramberg – Bäcklund , via un intermédiaire sulfone à cycle à trois chaînons.
Synthèse à partir de composés carbonylés
Une autre méthode importante pour la synthèse d’alcène implique la construction d’une nouvelle double liaison carbone-carbone par couplage d’un composé carbonyle (tel qu’un aldéhyde ou une cétone ) à un équivalent carbanion . Ces réactions sont parfois appelées oléfinations . La plus connue de ces méthodes est la réaction de Wittig , mais d’autres méthodes apparentées sont connues, notamment la réaction de Horner-Wadsworth-Emmons .
La réaction de Wittig implique la réaction d’un aldéhyde ou d’une cétone avec un réactif de Wittig (ou phosphorane) du type Ph 3 P=CHR pour produire un alcène et Ph 3 P=O . Le réactif de Wittig se prépare lui-même facilement à partir de triphénylphosphine et d’un halogénure d’alkyle. La réaction est assez générale et de nombreux groupes fonctionnels sont tolérés, même des esters, comme dans cet exemple : [19]

L’ oléfination de Peterson , qui utilise des réactifs à base de silicium à la place du phosphorane , est liée à la réaction de Wittig . Cette réaction permet la sélection des produits E – ou Z – . Si un produit E est souhaité, une autre alternative est l’ oléfination de Julia , qui utilise le carbanion généré à partir d’une phénylsulfone . L’ oléfination Takai basée sur un intermédiaire organochrome délivre également des E-produits. Un composé de titane, le réactif de Tebbe , est utile pour la synthèse de composés de méthylène ; dans ce cas, même les esters et les amides réagissent.
Une paire de cétones ou d’aldéhydes peut être désoxygénée pour générer un alcène. Les alcènes symétriques peuvent être préparés à partir d’un seul couplage aldéhyde ou cétone avec lui-même, en utilisant la réduction du titane métallique (la réaction de McMurry ). Si différentes cétones doivent être couplées, une méthode plus compliquée est nécessaire, telle que la réaction de Barton-Kellogg .
Une seule cétone peut également être convertie en alcène correspondant via sa tosylhydrazone, en utilisant du méthylate de sodium (la réaction de Bamford-Stevens ) ou un alkyllithium (la réaction de Shapiro ).
Synthèse à partir d’alcènes
La formation d’alcènes plus longs via la polymérisation par étapes de plus petits est attrayante, car l’éthylène (le plus petit alcène) est à la fois peu coûteux et facilement disponible, avec des centaines de millions de tonnes produites chaque année. Le procédé Ziegler-Natta permet la formation de très longues chaînes, par exemple celles utilisées pour le polyéthylène . Lorsque des chaînes plus courtes sont souhaitées, comme pour la production de tensioactifs , les procédés incorporant une étape de métathèse des oléfines , tels que le procédé Shell pour les oléfines supérieures, sont importants.
La métathèse d’oléfine est également utilisée commercialement pour l’interconversion de l’éthylène et du 2-butène en Propylène. Des catalyseurs hétérogènes contenant du rhénium et du molybdène sont utilisés dans ce procédé : [20]
CH 2 =CH 2 + CH 3 CH=CHCH 3 → 2 CH 2 =CHCH 3
L’ hydrovinylation catalysée par un métal de transition est un autre processus important de synthèse d’alcène à partir de l’alcène lui-même. [21] Cela implique l’addition d’un hydrogène et d’un groupe vinyle (ou d’un groupe alcényle) à travers une double liaison.
Des alcynes
La réduction des alcynes est une méthode utile pour la synthèse stéréosélective d’alcènes disubstitués. Si le cis -alcène est souhaité, l’hydrogénation en présence du Catalyseur de Lindlar (un Catalyseur hétérogène constitué de palladium déposé sur du carbonate de calcium et traité avec diverses formes de plomb) est couramment utilisée, bien que l’hydroboration suivie d’une hydrolyse fournisse une approche alternative. La réduction de l’alcyne par le sodium métallique dans l’ammoniac liquide donne le trans – alcène. [22]
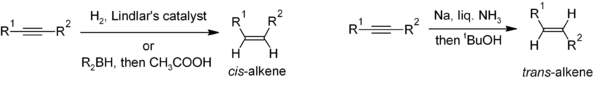
Pour la préparation d’alcènes multisubstitués, la carbométallation des alcynes peut donner lieu à une grande variété de dérivés d’alcènes.
Réarrangements et réactions associées
Les alcènes peuvent être synthétisés à partir d’autres alcènes via des réactions de réarrangement . Outre la métathèse des oléfines (décrite ci- dessus ), de nombreuses réactions péricycliques peuvent être utilisées telles que la réaction ène et le réarrangement de Cope .
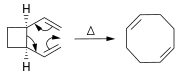
Dans la réaction de Diels-Alder , un dérivé de cyclohexène est préparé à partir d’un diène et d’un alcène réactif ou déficient en électrons.
Nomenclature IUPAC
Bien que la nomenclature ne soit pas largement suivie, selon l’IUPAC, un alcène est un hydrocarbure acyclique avec une seule double liaison entre les atomes de carbone. [3] Les oléfines comprennent une plus grande collection d’alcènes cycliques et acycliques ainsi que des diènes et des polyènes. [4]
Pour former la racine des noms IUPAC pour les alcènes à chaîne droite, remplacez l’ infixe -an- du parent par -en- . Par exemple, CH 3 -CH 3 est l’ alcane éthANe . Le nom de CH 2 =CH 2 est donc ethENe .
Pour les alcènes à chaîne droite avec 4 atomes de carbone ou plus, ce nom n’identifie pas complètement le composé. Dans ces cas, et pour les alcènes acycliques ramifiés, les règles suivantes s’appliquent :
- Trouvez la chaîne carbonée la plus longue de la molécule. Si cette chaîne ne contient pas la double liaison, nommez le composé selon les règles de dénomination des alcanes. Autrement:
- Numérotez les carbones de cette chaîne en commençant par l’extrémité la plus proche de la double liaison.
- Définissons l’emplacement k de la double liaison comme étant le numéro de son premier carbone.
- Nommez les groupes latéraux (autres que l’hydrogène) selon les règles appropriées.
- Définissez la position de chaque groupe latéral comme le numéro du carbone de la chaîne auquel il est attaché.
- Écrivez la position et le nom de chaque groupe latéral.
- Écrivez les noms des alcanes de même chaîne en remplaçant le suffixe « -ane » par « k -ène ».
La position de la double liaison est souvent insérée avant le nom de la chaîne (par exemple “2-pentène”), plutôt qu’avant le suffixe (“pent-2-ène”).
Les positions n’ont pas besoin d’être indiquées si elles sont uniques. A noter que la double liaison peut impliquer une numérotation des chaînes différente de celle utilisée pour l’alcane correspondant : (H3C)3C– CH2–CH _3est “2,2-diméthylpentane”, alors que (H3C)3C– CH = CH2est “3,3-diméthyl 1-pentène”.
Des règles plus complexes s’appliquent aux polyènes et aux cycloalcènes . [5]
 Nommer les hex-1-ènes substitués
Nommer les hex-1-ènes substitués
Cis – isomérie trans
Si la double liaison d’un mono-ène acyclique n’est pas la première liaison de la chaîne, le nom tel que construit ci-dessus n’identifie toujours pas complètement le composé, à cause de l’ isomérie cis – trans . Ensuite, il faut spécifier si les deux liaisons simples C – C adjacentes à la double liaison sont du même côté de son plan ou de côtés opposés. Pour les monoalcènes, la configuration est souvent indiquée par les préfixes cis – (du latin “de ce côté de”) ou trans – (“à travers”, “de l’autre côté de”) avant le nom, respectivement; comme dans le cis -2-pentène ou le trans –2-butène.
![]()
![]() La différence entre les isomères cis et trans
La différence entre les isomères cis et trans
Plus généralement, l’ isomérie cis – trans existera si chacun des deux carbones de la double liaison a deux atomes ou groupes différents qui lui sont attachés. En tenant compte de ces cas, l’IUPAC recommande la notation E–Z plus générale , au lieu des préfixes cis et trans . Cette notation considère le groupe avec la priorité CIP la plus élevée dans chacun des deux carbones. Si ces deux groupes sont sur les côtés opposés du plan de la double liaison, la configuration est étiquetée E (de l’ entgegen allemand signifiant « opposé »); s’ils sont du même côté, il est étiqueté Z (de l’allemand zusammen, “ensemble”). Cet étiquetage peut être enseigné avec la mnémonique ” Z signifie ‘on ze zame zide'”. [23]
 La différence entre les isomères E et Z
La différence entre les isomères E et Z
Groupes contenant des doubles liaisons C=C
L’IUPAC reconnaît deux noms pour les groupes hydrocarbonés contenant des doubles liaisons carbone-carbone, le groupe vinyle et le groupe allyle . [5]

Voir également
| Recherchez alcène dans Wiktionary, le dictionnaire gratuit. |
| Wikiquote a des citations liées à Alkene . |
- Alpha-oléfine
- Annulène
- Hydrocarbure Aromatique (“Arène”)
- Dendralène
- Nitroalcène
- Radialène
Liens nomenclatures
- Règle A-3. Composés insaturés et radicaux univalents [1] Livre bleu IUPAC.
- Règle A-4. Radicaux bivalents et multivalents [2] Livre bleu IUPAC.
- Règles A-11.3, A-11.4, A-11.5 Hydrocarbures monocycliques insaturés et substituants [3] Livre bleu IUPAC.
- Règle A-23. Composés hydrogénés d’hydrocarbures polycycliques fondus [4] Livre bleu IUPAC.
Références
- ^ un bc Wade , LG (2006). Chimie organique (6e éd.). Salle Pearson Prentice . p. 279 . ISBN 978-1-4058-5345-3.
- ^ H. Stephen Stoker (2015): Chimie générale, organique et biologique . 1232 pages. ISBN 9781305686182
- ^ un b IUPAC , Compendium de Terminologie Chimique , 2ème rédacteur. (le “Livre d’or”) (1997). Version corrigée en ligne : (2006–) ” alcènes “. doi : 10.1351/goldbook.A00224
- ^ un bc IUPAC , Compendium de Terminologie Chimique , 2ème rédacteur. (le “Livre d’or”) (1997). Version corrigée en ligne : (2006–) « oléfines ». doi : 10.1351/goldbook.O04281
- ^ un bc Moss , généraliste; Smith, PAS ; En ligneTavernier, D. (1995). “Glossaire des noms de classe des composés organiques et des intermédiaires réactifs basés sur la structure (recommandations IUPAC 1995)”. Chimie pure et appliquée . 67 (8–9) : 1307–1375. doi : 10.1351/pac199567081307 . S2CID 95004254 .
- ^ “Production : la croissance est la norme”. Nouvelles de la chimie et de l’ingénierie . 84 (28): 59-236. 10 juillet 2006. doi : 10.1021/cen-v084n034.p059 .
- ^ Sloane, NJA (éd.). “Séquence A000631 (Nombre de dérivés de l’éthylène à n atomes de carbone)” . L’ encyclopédie en ligne des séquences entières . Fondation OEIS.
- ^ Smith, Michael B.; March, Jerry (2007), Chimie organique avancée : réactions, mécanismes et structure (6e éd.), New York : Wiley-Interscience, p. 23, ISBN 978-0-471-72091-1
- ^ un b Bansal, Raj K. (1998). “Règle de Bredt” . Mécanismes de réaction organique (3e éd.). Éducation McGraw-Hill . p. 14–16. ISBN 978-0-07-462083-0.
- ^ Fawcett, Frank S. (1950). “La règle de Bredt des doubles liaisons dans les structures à anneaux atomiques pontés”. Chim. Rév. 47 (2): 219–274. doi : 10.1021/cr60147a003 . PMID 24538877 .
- ^ “La règle de Bredt”. Réactions et réactifs de noms organiques complets . Vol. 116. 2010. pp. 525–528. doi : 10.1002/9780470638859.conrr116 . ISBN 978-0-470-63885-9.
- ^ Duan, Xufang; Bloc, Éric ; Li, Zhen; Connelly, Timothée ; Zhang, Jian; Huang, Zhimin; Su, Xubo ; Casserole, Yi ; Wu, Lifang (28 février 2012). “Rôle crucial du cuivre dans la détection des odorants métal-coordination” . Actes de l’Académie nationale des sciences des États-Unis d’Amérique . 109 (9): 3492–3497. Bibcode : 2012PNAS..109.3492D . doi : 10.1073/pnas.1111297109 . ISSN 0027-8424 . PMC 3295281 . PMID 22328155 .
- ^ Streiwieser, A. ; Heathcock, CH ; Kosower, EM (1992). “11.6.G. Alcènes : réactions : ajouts de radicaux libres”. Introduction à la chimie organique (4e éd.). New York : Macmillan. p. 288.
- ^ Baptista, Mauricio S.; Cadet, Jean; Mascio, Paolo Di; Ghogare, Ashwini A.; Greer, Alexandre; Hamblin, Michael R.; Lorente, Caroline ; Nunez, Silvia Cristina; Ribeiro, Martha Simões; Thomas, Andrés H.; Vignoni, Marianne; Yoshimura, Tania Mateus (2017). “Réactions d’oxydation photosensibilisées de type I et de type II : lignes directrices et voies mécanistes” . Photochimie et photobiologie . 93 (4): 912–919. doi : 10.1111/php.12716 . PMC 5500392 . PMID 28084040 .
- ^ Wade, LG (2006). Chimie organique (6e éd.). Salle Pearson Prentice . p. 309 . ISBN 978-1-4058-5345-3.
- ^ Saunders, WH (1964). Patai, Saul (éd.). La Chimie des Alcènes . Wiley Interscience. p. 149–150.
- ^ Cram, DJ; Greene, Frederick D.; Depuy, CH (1956). “Études en stéréochimie. XXV. Effets d’éclipse dans la réaction E21”. Journal de l’American Chemical Society . 78 (4): 790–796. doi : 10.1021/ja01585a024 .
- ^ Bach, RD; Andrzejewski, Denis; Dusold, Laurence R. (1973). “Mécanisme de l’élimination de Cope”. J. Org. Chim . 38 (9) : 1742–3. doi : 10.1021/jo00949a029 .
- ^ Snider, Barry B.; Matsuo, Y; Snider, BB (2006). “Synthèse d’ent-Thallusin” . Org. Lett . 8 (10): 2123–6. doi : 10.1021/ol0605777 . PMC 2518398 . PMID 16671797 .
- ^ Lionel Delaude, Alfred F. Noëls (2005). “Métathèse”. Encyclopédie Kirk-Othmer de la technologie chimique . Weinheim : Wiley-VCH. doi : 10.1002/0471238961.metanoel.a01 . ISBN 978-0471238966.{{cite encyclopedia}}: Maint CS1 : utilise le paramètre auteurs ( lien )
- ^ Vogt, D. (2010). “Hydrovinylation asymétrique catalysée au cobalt”. Angew. Chim. Int. Éd . 49 (40): 7166–8. doi : 10.1002/anie.201003133 . PMID 20672269 .
- ^ Zweifel, George S.; En ligneNantz, Michael H. (2007). Synthèse Organique Moderne : Une Introduction . New York : WH Freeman & Co. p. 366 . ISBN 978-0-7167-7266-8.
- ^ John E. McMurry (2014): Chimie organique avec applications biologiques ; 3e édition. 1224pages. ISBN 9781285842912